病理学论文

摘 要: 内质网应激(endoplasmic reticulum stress,ERS)是真核细胞普遍存在的应激–防御机制。ERS状态下,细胞会启动未折叠蛋白反应(unfolded protein response,UPR)增强对未折叠蛋白的折叠和对错误折叠蛋白的降解,以恢复内质网的正常生理功能。一些引发ERS的刺激也会诱发细胞自噬。自噬作为真核细胞保守的降解机制,可通过加快错误折叠蛋白的降解,降低ERS水平,是继UPR之外帮助内质网恢复稳态的另一重要角色。研究表明,ERS及其伴随的细胞自噬与很多疾病的发生发展密切相关。然而,ERS如何引发细胞自噬,自噬如何反馈调节ERS,UPR与细胞自噬如何关联,这些问题并未得到详细的探讨和阐释。因此,该文对ERS和细胞自噬的关系及其关联机制进行综述,以期为相关疾病发病机制的阐明和开发新的治疗策略提供依据。
关键词: 内质网应激; 未折叠蛋白反应; 自噬;
Abstract: ERS (endoplasmic reticulum stress) is a universal stress-defense mechanism in eukaryotic cells.When ERS appears,UPR (unfolded protein response) is initiated,enhancing the unfolded proteins folding and misfolded proteins degradation,to restore normal physiological functions of ER.Some stimuli that trigger ERS can also induce autophagy.As a conservative degradation mechanism of eukaryotic cells,autophagy can increase the degradation of misfolded proteins and reduce the ERS,playing an important role on the maintenance of ER homeostasis besides UPR.Studies have demonstrated that ERS and ERS-induced autophagy are closely related to the occurrence and development of many diseases.However,how ERS triggers autophagy,how autophagy feedback regulates ERS,how UPR correlates autophagy and the roles of regulatory mechanisms between each other in the development of related diseases have not been discussed and defined in detail.The comprehensive view of the correlated mechanism between ERS and autophagy is significant for elucidating the pathogenesis of related diseases.Therefore,this article reviews the relationship and regulatory mechanism between ERS and autophagy in order to provide new ideas for the research of related fields.
Keyword: endoplasmic reticulum stress; unfolded protein response; autophagy;

内质网是真核细胞内重要的膜性细胞器,在蛋白质的合成、加工、运输,脂质的生物合成,Ca2+平衡的调节等方面发挥重要作用[1]。内质网是细胞内膜系统中蛋白质监控的一个重要环节,正确折叠的蛋白质被运输出内质网腔,错误折叠的蛋白质被保留在内质网中,最终被降解。内质网腔具有高Ca2+浓度和强氧化环境两个重要特征,前者使内质网能够调节细胞内钙稳态,后者为蛋白质合成、折叠、运输等提供必要条件[2]。稳定的内质网环境是实现内质网功能的基本条件。内质网对内外界刺激特别敏感,如氧化应激、Ca2+失衡、缺氧、缺糖、炎症刺激等都会造成内质网腔的强氧化环境被破坏,导致内质网功能出现障碍,产生突变的蛋白质或者蛋白质二硫键不能形成,引起内质网中未折叠或错误折叠蛋白的积聚,诱发内质网应激(endoplasmic reticulum stress,ERS)。细胞为了消除内质网中积聚的蛋白启动未折叠蛋白反应(unfolded protein response,UPR)[3]。UPR具有双重性,UPR的激活可以维持细胞存活,但在严重或持续的ERS条件下,UPR的适应性反应不足以消除ERS,细胞不能恢复内质网稳态,最终引发细胞凋亡[4,5]。细胞自噬是伴随UPR发生的另一种细胞保护机制,帮助降解内质网中积聚的未折叠或错误折叠蛋白,降低ERS,维持内质网稳定性,保护细胞存活。研究表明,在多种生理与病理过程中,ERS与自噬存在相互作用。下文将对ERS和细胞自噬关系的研究进展加以概述,为ERS、自噬及相关疾病关系的研究提供理论指导。
1、 ERS与UPR
当ERS持续较长时间时,内质网功能紊乱,细胞激活UPR信号通路,以应对环境变化,恢复内质网利于蛋白质折叠的环境,维持细胞内环境的稳定,是细胞的一种自我保护机制。UPR主要通过减少蛋白质的合成、增强蛋白质的折叠能力、加快错误折叠蛋白质的降解来重建内质网稳态。在ERS的早期阶段,细胞通过调控转录、翻译和翻译后过程减缓新生蛋白流入内质网,同时上调与内质网蛋白降解相关的蛋白表达,提高内质网对蛋白质的折叠加工能力和对错误折叠蛋白的降解能力,以逆转ERS。若ERS不能逆转,UPR会激活细胞自噬甚至诱发细胞凋亡,以实现内环境的稳定[6]。
哺乳动物细胞内UPR由三种内质网跨膜感受器介导:双链RNA依赖的蛋白激酶样内质网激酶(RNA dependent protein kinase-like ER kinase,PERK)、肌醇需酶1α(inositol requiring enzyme 1α,IRE1α)和活化转录因子6(activating transcription factor 6,ATF6),三种感受器并行工作,激活各自的信号转导途径[7]。在正常的生理状态下,PERK、IRE1α、ATF6均与葡萄糖调节蛋白78(glucose regulated protein 78,GRP78)结合处于非活性状态[8]。在ERS条件下,内质网内大量积聚的错误折叠或未折叠蛋白优先与GRP78结合,导致GRP78与三种感受器分离,激活UPR信号转导途径,促进蛋白质正确折叠和错误折叠蛋白降解,以恢复内质网环境的稳定[9]。GRP78是热休克蛋白Hsp70(heat shock protein 70)家族的成员之一,是一种钙结合蛋白,一方面,它作为分子伴侣介导新生蛋白质的正确折叠和装配,并协助蛋白质跨内质网膜转运;另一方面,它也是内质网的一种应激蛋白,与内质网中未折叠蛋白的积累密切相关。在发生ERS时,GRP78的表达量显着增高,通过转移内质网腔内的错误折叠蛋白,保证细胞在应激状态下蛋白质合成的继续,帮助维持内质网钙稳态及内环境稳定。GRP78是内质网稳态的感受器,被认为是ERS的标志蛋白质[10,11]。
2、细胞自噬
2.1、自噬的类型
细胞自噬是真核生物进化过程中高度保守的一类降解途径,通过形成双层膜的封闭囊泡,即自噬体(autophagosome),将蛋白质等生物大分子、线粒体等细胞器以及微生物等回收至溶酶体形成自噬溶酶体(autolysosome),将回收的物质降解为氨基酸、单糖等小分子,实现物质循环再利用的动态过程。这是研究者通常所说的自噬,也是目前研究最为广泛和深入的自噬种类,即巨自噬(macroautophagy)。根据降解底物进入溶酶体的方式分类,自噬还有微自噬(microautophagy)和分子伴侣介导的自噬(chaperone-mediated autophagy,CMA)两种形式[12]。根据降解底物的特异性,自噬还可以分为选择性自噬和非选择性自噬[13]。常见的选择性自噬有线粒体自噬、内质网自噬(ER-phagy)和过氧化物酶体自噬等[13]。目前大部分研究认为,自噬的发生起始于内质网上呈杯型结构的欧米茄体(omegasome)—一个招募自噬因子的平台,在omegasome里衍生出双层膜的自噬前体(phagophore),phagophore在自噬因子的作用下不断富集磷脂分子得以生长弯曲,逐渐延伸为能够部分包裹待降解底物的新月形结构,随着双层膜的不断延展最后闭合成为封闭的囊泡即自噬体(autophagosome),之后autophagosome携带底物与溶酶体融合为autolysosome,底物在溶酶体酶的作用下被消化分解为小分子物质,供细胞再次利用[12,14]。
细胞自噬是细胞在饥饿条件下的一种存活机制。在营养充足的条件下,自噬发生处于较低水平,仅在胞质中去除受损的细胞器和积聚的蛋白质等以维持细胞内环境稳定,称为基础自噬[12]。当营养缺失时,细胞自噬大量启动以维持胞质中氨基酸池的平衡,通过合成新的蛋白质、能量生成、促进糖异生来避免细胞“饿死”,称为诱导自噬[12]。越来越多的研究发现,自噬不仅是细胞感知能量缺乏和应激条件的适应性反应,而且参与许多重要的生理过程,自噬与组织器官发育[15]、个体衰老[16]、 炎症[17]、外源微生物的清除[18]、细胞免疫[17]、肿瘤[19]和神经退行性疾病[20]等的发生等密切相关。
2.2、自噬的调控
自噬对维持细胞、组织乃至器官的稳态至关重要。该过程受自噬相关基因(autophagy-related genes,ATGs)和蛋白激酶的严密调控。mTOR(mammalian target of rapamycin)被认为是控制细胞自噬的中心分子,它通过调控自噬起始分子ATG1/ULK1(Unc-51-like kinase)激酶复合体[包括ULK1、FIP200(FAK-family interacting protein of 200 kDa)、ATG13和ATG101]的活性来调控自噬。mTOR本身是一个调控细胞周期、生长和增殖的丝/苏氨酸激酶,正常情况下,mTOR处于激活状态,磷酸化ULK1、ATG13,抑制ULK1激酶复合物的形成,抑制ULK1的活性,自噬被抑制[21]。当细胞感受到压力如饥饿、生长因子缺乏等,mTOR活性被抑制,其对ULK1激酶复合体的抑制解除,ATG13与ULK1、ATG101和FIP200结合成为有活性的ULK1激酶复合体,自噬启动[22]。AMPK(AMP-activated protein kinase)是细胞中感受能量状态的蛋白激酶,在自噬发生的调控中也发挥重要作用。在能量匮乏时,AMPK活化,一方面通过磷酸化TSC(tuberous sclerosis proteins)1/2复合物和mTOR的特异组分Raptor,抑制mTOR的活性;另一方面通过磷酸化ULK1,直接激活ULK1[23],诱导自噬的起始。
ULK1激活后移位到内质网凹陷处即omegasome的产生部位,磷酸化Beclin-1、ATG14,促进Beclin-1/PI3KC3(phosphoinositide-3-kinase class 3)复合物[包括Beclin-1、VPS34(vacuolar sorting protein 34)、p150和ATG14]的形成,提高VPS34的脂激酶活性[24],并协助该复合物移位到omegasome中,由ATG14结合内质网膜将复合物锚定[25]。VPS34在此催化内质网合成的磷脂酰肌醇生成3-磷酸磷脂酰肌醇(phosphatidylinositol 3-phosphate,PI3P),PI3P分子在omegasome里聚集成脂库,不断招募PI3P结合因子,如含FYVE、WD40重复序列、 PX结构域的蛋白因子[26,27],帮助phagophore延伸,其中WIPI[WD-repeat PtdIns(3)P effector protein]家族蛋白在自噬体膜的延展生长中发挥了很重要的作用。另外,两个类泛素化系统:ATG5-ATG12-ATG16L连接系统和ATG8/LC3连接系统直接参与了自噬体膜的延伸[28]。Beclin-1/PI3KC3复合物产生的PI3P广泛招募WIPI2蛋白到phagophore上,WIPI2能够与ATG16L相互作用[29,30],将下游类泛素化系统ATG5-ATG12-ATG16L复合体也富集到phagophore上,该复合体作为LC3的E3连接酶,帮助LC3连接磷脂酰乙醇胺(phosphatidylethanolamine,PE)分子,形成具有膜结合能力的LC3-II[31],随着酯化形式的LC3-II分子不断整合到phagophore的双层膜上,新生的自噬体膜得以扩展延伸,自噬体逐渐成熟。研究发现,phagophore的生长和闭合也与ATG2-WIPI4复合物有关,在LKB1介导的AMPK信号激活后,WIPI4-ATG2复合物从WIPI4-ATG2/AMPK-ULK1复合体上释放,移位到初生自噬体phagophore上,帮助其生长变大[32]。自噬体从phagophore生长为成熟的autophagosome的过程及其所需的关键蛋白因子如图1所示。
图1自噬体形成过程(根据参考文献[33]修改)
![图1自噬体形成过程(根据参考文献[33]修改)](http://www.xueshut.com/uploads/allimg/201205/36-201205092159595.jpg)
Fig.1 Process of autophagosome formation (modified from reference[33])
另外,乙酰化和泛素化修饰也是自噬调控的重要方式。在营养充足时,细胞内的LC3、ATG5、ATG7和Beclin-1/VPS34等自噬相关蛋白被乙酰化酶p300乙酰化[34,35],LC3分子被局限于细胞核,不能进入细胞质参与自噬[36,37],同时Beclin-1/VPS34的活性被抑制,自噬不能启动。氨基酸缺乏时,mTORC1失活,不能激活p300,p300的活性被抑制[38],LC3等分子去乙酰化加强,LC3进入细胞质,Beclin-1/VPS34去乙酰化恢复活性[39],自噬启动。p300-VPS34途径除了在营养缺乏诱导的经典细胞自噬中发挥作用外,对不依赖于VPS34上游信号分子(AMPK、mTORC1或ULK1)的非经典自噬的启动也很重要[39]。此外,mTORC1可通过直接磷酸化ATG14,抑制Beclin-1/VPS34的活性,抑制自噬体膜的成核[40],还可直接磷酸化WIPI2蛋白控制它的泛素化降解,影响自噬体膜的延伸[41]。另外,ULK1、Beclin-1、Bcl-2(B-cell lymphoma-2)的泛素化也影响相关复合物的活性进而影响自噬的启动[42]。自噬溶酶体完成底物的降解后,生成的小分子氨基酸重新激活mTOR,活化的mTOR一方面抑制自噬的发生,另一方面通过激活PIK3C3-UVRAG(UV irradiation resistance-associated gene)复合体启动autolysosome的管状化和片段化,促进溶酶体的再生,实现细胞器的更新[43]。可见,mTOR是经典自噬途径调控的关键,在整个自噬的发生包括自噬的启动、自噬体生长成熟乃至自噬降解完成后溶酶体的再生阶段都发挥调控作用,mTOR通过对细胞内自噬体和溶酶体机器的精准调节,实现细胞内物质代谢的平衡和稳定。
3、 ERS与细胞自噬的相互作用
3.1、 ERS诱导细胞自噬
细胞自噬和ERS是两个独立的生物过程,越来越多的研究表明,二者存在相互作用,ERS可引发细胞自噬,自噬通过降解积聚的错误折叠蛋白质来减轻内质网的压力。早在20世纪80年代,HORNUNG等[44]通过超微结构研究发现,具有自噬泡的细胞通常伴有内质网扩张现象。自噬是细胞营养缺乏时的反应,但有研究发现,使用ERS诱导剂可诱导酵母细胞在营养充足的条件下发生自噬,说明ERS可以激活细胞自噬[45]。持续发生的ERS通过UPR激活细胞自噬,降解错误折叠蛋白,恢复内质网稳态,促进细胞存活[46]。GRP78作为ERS标志蛋白,对于ERS诱导自噬发生非常重要,研究发现,在GRP78表达被敲低的细胞中,内质网功能失常,尽管UPR途径和LC3的酯化仍可进行,但ERS和营养缺乏诱导的自噬体的形成受到抑制[47]。可见,ERS需要自噬作为保护机制,同时自噬的完成依赖于ERS。ERS与细胞自噬的适度激活形成了机体适应性、保护性的交互作用方式。
3.2、 UPR与细胞自噬
UPR可降低未折叠或错误折叠蛋白的生成,增强内质网对蛋白质的处理能力,恢复内质网稳态。当ERS持续发生时,UPR通过PERK、IRE1α、ATF6三种感受器激活各自的信号转导通路,并进一步激活或者上调自噬关键分子,诱导细胞发生自噬,加强对错误折叠蛋白的降解以减轻ERS压力,保护细胞存活。
3.2.1、PERK信号通路与细胞自噬
PERK-eIF2α (eukaryotic initiation factor 2α)通路是介导ERS和细胞自噬之间相互作用的重要调控通路之一。PERK是内质网I型跨膜蛋白,具有丝/苏氨酸蛋白激酶活性,属于eIF2α蛋白激酶家族成员。PERK的二聚化结构域可感知ERS信号。在正常生理条件下,二聚化位点与GRP78结合而被覆盖。当ERS发生时,GRP78优先与内质网内积聚的错误折叠或未折叠蛋白结合,导致GRP78与PERK分离,PERK通过二聚化和自身磷酸化被激活。活化的PERK特异地磷酸化eIF2α,抑制其翻译起始活性,减少蛋白质的翻译抑制蛋白质合成,从而降低内质网负荷[48]。ERS发生时,PERK磷酸化eIF2α,会使很多蛋白翻译被抑制,而有些蛋白却恰恰需要eIF2α的磷酸化,如应激特异性转录因子4(activating transcription factor 4,ATF4)的激活。ATF4激活后诱导一系列基因表达促进UPR如GRP78等分子伴侣和蛋白质转运相关基因的表达,增强细胞对蛋白质的折叠和转运能力,减少蛋白质聚积,恢复内质网稳态。
在细胞自噬过程中,两个相互依赖的泛素样结合系统即ATG12-ATG5连接系统、ATG8(LC3)酯化系统起着极为重要的作用。ERS负担过重时,PERK通过ATF4激活自噬启动因子Beclin-1、泛素样蛋白系统LC3、ATG5、ATG7、ATG12和ATG16L的表达[49]、以及参与泛素化底物特异性降解的p62和NBR1(neighbor of BRCA1)的表达[50],促进细胞自噬发生,加强对内质网聚集蛋白的降解,辅助UPR减轻ERS,从而维持细胞在长时间应激状态下的生存。多聚谷氨酰胺Q72(polyglutamine Q72)的异常表达和积聚会导致ERS和细胞自噬,由PERK-eIF2α-ATF4途径介导ATG12、LC3基因的上调以及LC3的酯化,促进细胞自噬的发生[51]。PERK-eIF2α-ATF4途径作为自噬相关蛋白库的补充途径,在维持细胞自噬状态方面可能起重要作用[52,53]。引发该通路的ERS刺激和具体的生理病理环境目前并不十分明晰,有待于进一步阐明。C/EBP同源蛋白(C/EBP-homologous protein,CHOP)是ERS时表达升高的另一个标志蛋白,其表达也受ATF4转录因子调控。研究表明,CHOP在ERS触发的细胞自噬中发挥重要作用,如缺氧诱导的ERS中,CHOP可上调自噬基因ATG5的表达,促进自噬体的形成[54,55]。抗结直肠癌药物牛樟芝诱导的结直肠癌细胞ERS中,CHOP激活下游基因TRB3(tribbles pseudokinase 3)的表达,TRB3与AKT直接结合抑制其磷酸化,阻碍AKT对mTOR的激活,活化ULK1引发自噬,但诱导的自噬最终导致结直肠癌细胞死亡[56],在此,CHOP-TRB3-AKT-mTOR介导了自噬性细胞死亡,可作为一种新的抗癌机制,同时也是ERS无法消除时的一种机体保护机制。另有研究表明,CHOP可通过激活生长停滞及DNA损害可诱导蛋白34(growth arrest and DNA damage-inducible gene 34,GADD34)促进elF2α蛋白去磷酸化以提高蛋白翻译水平,加重细胞内蛋白质负荷,诱导细胞凋亡[57]。此外,CHOP还可诱导Bcl-2家族抗凋亡蛋白的表达下调和促凋亡蛋白Bax/Bak(Bcl-2 associated x/k protein)的表达上调,促进细胞凋亡[58]。ERS时,CHOP介导的细胞死亡究竟是自噬性死亡还是凋亡?这可能与细胞受到的刺激类型以及强度有关,有待进一步研究。
3.2.2、 IRE1α信号通路与细胞自噬
IRE1α是内质网I型跨膜蛋白,具有激酶和核酸内切酶活性。正常生理条件下,IRE1α与GRP78结合处于非活性状态,ERS发生时,GRP78与IRE1α分离,IRE1α自身发生磷酸化而被激活,激活的IRE1α与肿瘤坏死因子受体相关因子2(tumor necrosis factor receptor-associated factor 2,TRAF2)和细胞凋亡信号调节激酶(apoptosis signal-regulating kinase,ASK)结合形成复合物,磷酸化c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)使其活化。活化的JNK磷酸化Bcl-2,将Beclin-1从Beclin-1/Bcl-2复合物中释放出来,并与III型磷脂酰肌醇激酶PI3KC3结合构成激酶复合物,参与自噬体的成核。此外,在JNK表达被敲低的细胞中,ATG7的表达也降低[59],这表明,JNK信号通路通过调控ATG7的表达调节自噬。在IRE1α缺失或经JNK抑制剂处理的细胞中,ERS诱导的自噬被抑制[60]。使用依赖IRE1α传感器的UPR诱导剂衣霉素和阿普西甘,能够驱动人神经母细胞瘤细胞系SK-N-SH细胞中自噬体的形成和小鼠胚胎成纤维细胞LC3酯化的增加[61]。以上研究表明,IRE1α-ASK-JNK通路介导了ERS和细胞自噬之间的正向调控。
IRE1α-XBP1(X-box binding protein 1)通路是ERS关联细胞自噬的第二个重要调控通路。IRE1α激活后具有了核酸内切酶活性,选择性切割UPR调节因子XBP1,激活其转录因子活性,上调与内质网蛋白折叠、转运、降解相关基因的转录,加强未折叠蛋白的折叠和错误折叠蛋白的降解[3],减轻或终止ERS,促进细胞内环境恢复稳态。在亨廷顿疾病小鼠中,伴随ERS和UPR出现,在条件性敲除XBP1蛋白后观察到自噬水平的升高,疾病的发展得到延缓,进一步研究发现,XBP1通过FoxO1(forkhead box O1)对自噬起负调控作用[62]。另有研究表明,在内皮细胞中,IRE1α-XBP1能够正向调控自噬,主要是通过Beclin-1的转录激活引发自噬[63]。ERS、XBP1和自噬也与肿瘤的发生发展密切相关,恶性肿瘤细胞的快速增殖和相对缺氧、营养缺乏等微环境均可诱导ERS[64]。乳腺癌[65]、结直肠癌等[66]恶性肿瘤中都观察到XBP1表达水平升高,沉默XBP1基因后,肿瘤血管生成减少,肿瘤生长减少且细胞凋亡增加[67]。而恶性肿瘤中自噬功能是相对缺陷的[68],恶性肿瘤中高表达的XBP1与缺陷的自噬功能之间是否也存在一定的负向调节,目前尚无确切证据,需要进一步探讨。目前对ERS相关疾病中IRE1α-XBP1信号通路与自噬的关联机制并不十分清楚,XBP1对细胞自噬的正/负向调控可能与细胞类型、细胞状态、细胞所处的环境有关,其中的具体机制需要更多深入的研究,以更好地解释具体疾病的发病机理,为ERS相关疾病的治疗和预后提供依据。
3.2.3、 ATF6信号通路与细胞自噬
ATF6是内质网II型跨膜蛋白,属于亮氨酸拉链家族bZIP(basic-region leucine zipper)结构域的转录因子。正常生理状态下,ATF6与GRP78结合处于非活性状态,在ERS状态下,GRP78与ATF6分离,ATF6易位至高尔基体,先后被位点-1丝氨酸蛋白酶(serine proteasesite-1,S1P)和位点-2金属蛋白酶(metalloprotease site-2 protease,S2P)水解而被激活,然后转入细胞核,与ERS反应元件结合,诱导相关的内质网伴侣蛋白表达,如GRP78、葡萄糖调节蛋白94(glucose regulated protein 94,GRP94)、内质网蛋白57(endoplasmic reticulum protein 57,ERp57)以及内质网蛋白降解相关的蛋白表达[1],促进未折叠蛋白正确折叠,加快错误折叠蛋白降解,促进细胞内环境恢复稳态。
ERS负担过重时,ATF6激活死亡相关蛋白激酶(death-associated protein kinase,DAPK),其是一种钙调蛋白调节丝/苏氨酸蛋白激酶,活化的DAPK磷酸化Beclin-1,将Beclin-1从Beclin-1/Bcl-2复合物中释放出来,激活自噬[69]。ATF6还可以通过上调GRP78的表达来抑制AKT Ser473位点磷酸化,进而抑制其下游mTOR的活性,激活自噬[70]。丙型肝炎病毒感染时,ATF6可上调DNA损伤诱导转录因子3(DNA damage-inducing transcription factor 3,DDIT3)的表达,DDIT3可与LC3基因启动子区结合,促进LC3的表达,进而促进细胞自噬[71]。目前,ATF6通路在ERS诱导自噬中的研究尚少,需要更广泛深入的研究。
未折叠蛋白反应介导的细胞自噬,信号通路如图2所示。研究发现,ERS不会同时激活三条UPR信号通路,IRE1α通路和ATF6通路首先被激活,并且随着ERS的持续发生,IRE1α和ATF6的活性逐渐减弱;而包含促凋亡转录调节子CHOP的PERK通路后续被激活,并在ERS发生过程中持续活化,最终细胞走向死亡;当人工维持IRE1α的活性时,细胞存活率升高,表明不同UPR分支信号的持续时间是ERS发生时细胞生死的决定因素[72,73]。不同信号的持续时间不同,其中具体的分子机制有待进一步阐明。ERS发生时,UPR的三条信号通路首先是通过抑制错误蛋白的合成,加强内质网对非折叠蛋白的折叠能力以及加速错误折叠蛋白的降解来维持内质网稳态的,此时并不需要激活细胞自噬途径,适度的ERS实是细胞自身代偿和自身保护的过程。
3.3、其他可引起ERS的信号与细胞自噬
ERS可通过UPR通路对自噬直接进行调节,也可通过细胞内氧含量、ROS和Ca2+浓度等间接调节自噬。
3.3.1、低氧与细胞自噬
蛋白质折叠是一个需氧耗能过程,缺氧会降低蛋白质的折叠能力,导致内质网腔中积聚大量的未折叠或错误折叠蛋白,引发ERS。PERK-eIF2α-ATF4信号通路介导的自噬激活是细胞应对低氧刺激的主要适应机制。肿瘤细胞生存环境严重缺氧时,PERK途径中的ATF4和CHOP被激活,ATF4直接结合到LC3启动子区上调LC3的表达,CHOP上调ATG5的基因表达,促进自噬的发生[54,55]。另有研究发现,在低氧状态下,SK-N-SH神经母细胞瘤通过IRE1α募集TRAF2和ASK,磷酸化JNK使其活化,进而磷酸化Bcl-2,导致Bcl-2与Beclin-1解离,引发自噬,帮助肿瘤细胞存活。可见,低氧通过ERS诱导自噬的信号机制并不唯一,可能与具体的组织器官代谢方式及缺氧程度有关。
图2未折叠蛋白反应介导的细胞自噬
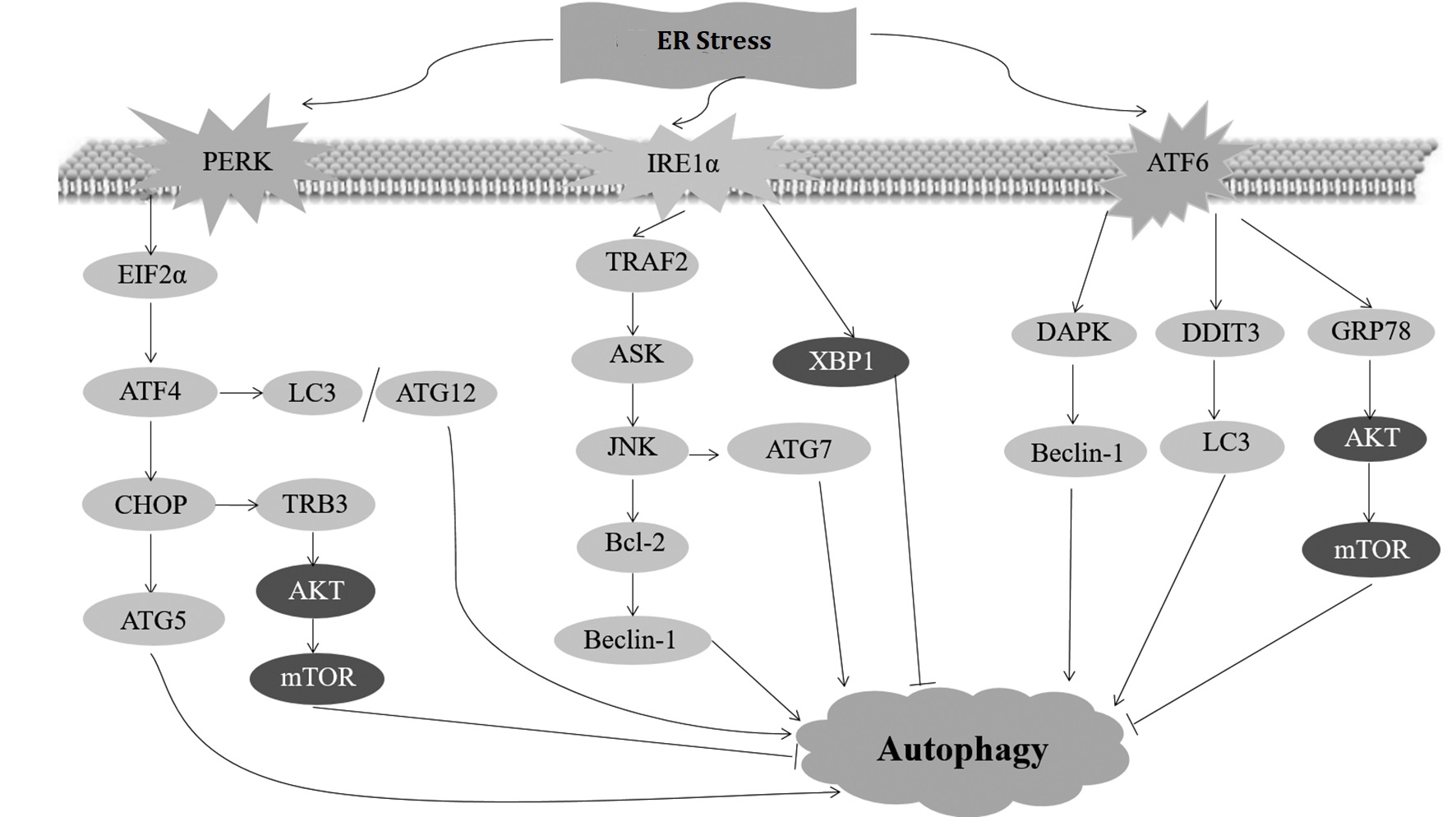
Fig.2 UPR-mediated autophagy
PERK介导的UPR可通过上调自噬相关基因LC3、ATG5、ATG12等的表达,促进LC3的酯化以及抑制mTOR活性促进自噬发生;IRE1α介导的UPR通过上调ATG7的表达以及从Bcl-2释放Beclin-1分子促进自噬的发生,亦可通过XBP1分子对自噬起负调控作用;ATF6介导的UPR可通过上调LC3的表达,释放Beclin-1分子以及抑制mTOR活性激活自噬
PERK-mediated UPR promotes autophagy by up-regulating the expression of autophagy-related genes such as LC3,ATG5,ATG12,etc,increasing LC3 lipidation and inhibiting mTOR activity.IRE1α-mediated UPR not only positively regulates autophagy by up-regulating ATG7 expression and releasing Beclin-1 from Bcl-2,but also negatively regulates autophagy through XBP1.ATF6-mediated UPR initiates autophagy by up-regulating LC3 expression,releasing Beclin-1,and inhibiting mTOR activity.
通常情况下,低氧诱导ERS激活的自噬对细胞起保护作用。自噬能够降解积聚的错误折叠蛋白来减轻ERS,同时,自噬回收降解受损细胞器和聚集的蛋白质为细胞生存提供营养,这非常有利于血管闭塞后细胞的存活[74]。研究发现,在严重缺氧情况下,ATF4诱导的自噬可防止乳腺癌细胞死亡[55]。但是也有研究表明,低氧诱导ERS激活的自噬对细胞具有损害作用,在缺氧状态下,适度的自噬对神经元的存活和功能至关重要,异常激活的自噬会对神经元造成损伤。在小鼠缺血缺氧模型中,LC3-II水平显着升高,海马神经元大量死亡,而自噬调节基因ATG7缺陷的小鼠海马神经元细胞的死亡减少[75]。然而,ERS诱导的自噬是否直接导致了神经元死亡,自噬对细胞生存和死亡的调控节点如何界定,目前并不清楚。这些不同的研究结果可能与自噬的诱导因素和细胞所处的环境有关。肿瘤组织由于其癌细胞高速增殖和代谢旺盛,细胞内是一个相对缺氧的环境,如何有效调控肿瘤细胞的ERS和自噬活性,使其不但能特异诱导肿瘤细胞死亡,同时又不损伤正常细胞?这将是利用自噬和低氧诱导的ERS进行肿瘤防治的关键。有待于更深入、具体的ERS激活自噬、UPR与自噬关联信号传导机制的挖掘和阐明。
3.3.2、氧化应激与细胞自噬
活性氧(reactive oxygen species,ROS)是细胞代谢过程中产生的氧的部分还原代谢产物,作为一种胞内信号分子参与细胞增殖、凋亡、免疫等生物过程。正常生理状态时,机体内ROS的产生与清除处于动态平衡,但在一些体内外刺激下,动态平衡被打破,ROS产生过量,机体无法及时清除,则发生氧化应激[76],进而触发ERS。研究发现,ROS能够通过原癌基因K-Ras激活JNK,上调自噬相关蛋白ATG5、ATG7的表达,引发细胞自噬[77]。自噬通过清除被ROS损伤的细胞器,降低ROS对细胞的损伤,保护细胞以利于细胞存活。但若细胞内ROS水平过高,超出自噬能够处理的范围,则细胞会因为自噬过强走向死亡[78],这种自噬导致的细胞死亡与自噬伴随的细胞凋亡是不同的两个概念,主要表现为细胞质被大量降解。ROS介导的自噬参与了动脉粥样硬化、阿尔茨海默病、帕金森病等疾病的发生发展,但其在具体疾病中的作用及机制仍需进一步阐明。未来针对自噬与ROS介导的ERS信号通路的精确调控,以及避免或减少其从保护机制转为损伤机制将为ROS相关疾病的防治产生重要影响。
3.3.3、 Ca2+与细胞自噬
Ca2+是细胞内一种重要的信号调节因子,参与细胞代谢、基因转录、分泌等各类细胞功能的调控。内质网是细胞的钙储存库,ERS会导致Ca2+从内质网释放到胞浆,诱导细胞自噬。CaMKK-β((calmodulin-depen-dent kinase kinase β)/AMPK/mTOR途径是Ca2+诱导细胞自噬的重要调节通路。首先,内质网释放的Ca2+激活CaMKK-β,进而激活AMPK,mTOR活性被抑制,ULK1活化形成复合体,诱发细胞自噬[79]。Ca2+还可以激活DAPK,活化的DAPK可使Beclin-1的BH3区域Thr119磷酸化,促使Beclin-1与Bcl-2解离,引发细胞自噬[80]。另有研究表明,胞质内Ca2+浓度的增加可以独立诱导自噬的发生,不需要通过AMPK活化和mTOR信号的抑制[81]。例如,短时间内用高浓度的Ca2+动员剂毒胡萝卜素处理细胞,使细胞处于急性ERS状态,内质网释放的Ca2+可激活蛋白激酶C(protein kinase C,PKC),通过不依赖mTOR的机制诱导自噬[82]。这些不同的研究结果说明,Ca2+诱导的自噬既存在经典自噬途径,也存在非经典自噬途径,可能与细胞的类型及细胞受到的环境刺激因素有关。
3.4、自噬对ERS的负反馈调节
自噬对ERS的调控多为抑制作用,通过加快错误折叠蛋白的降解缓解ERS水平,恢复内质网的正常生理功能。一项小鼠肾脏缺血再灌注的研究发现,在初始再灌注阶段,肾脏细胞出现ERS,ERS标志蛋白GRP78、PERK等表达增多,加入雷帕霉素进行再灌注,肾脏细胞自噬增加,ERS标志蛋白的表达降低,肾脏细胞的正常功能得以维持;而合并自噬抑制剂再灌注后,雷帕霉素对肾脏细胞的保护作用减弱[83],表明肾脏缺血初始再灌注阶段细胞自噬的激活可抑制ERS以修复肾脏细胞功能。另有研究表明,在诱导帕金森病的刺激因素下,CMA亦可抑制ERS,维持黑质致密部(substantia nigra pars compacta,SNc)多巴胺能神经元的生存,减缓帕金森病的进程。CMA途径受溶酶体相关膜蛋白2A(lysosome-associated membrane protein 2A,LAMP2A)和热激同源蛋白70(heat shock cognate protein 70,HSC70)的调控,ERS诱导剂能够通过PERK-MAPK14通路磷酸化LAMP2A使之活化,进而诱导CMA,降解积累的受损蛋白,降低ERS水平。内质网是细胞的钙储存库,内质网伴侣蛋白钙网蛋白(calreticulin,CRT)对于维持钙稳态起重要作用并参与自噬的调控。钙稳态失衡时,大量错误折叠或未折叠蛋白堆积在内质网,引发ERS和UPR,CRT表达上调,CRT通过其保守的LIR(LC3 interacting region)序列与LC3相互作用[84],促进细胞自噬,降解内质网内积聚的错误折叠蛋白,缓解细胞应激水平,促进细胞内环境恢复稳态,CRT-自噬途径介导了钙稳态失衡时对ERS的负反馈调节[85]。还有研究发现,ERS时,多种定位于内质网的受体蛋白参与ER-phagy以逆转ERS,维持细胞内稳态。如受体蛋白Sec62、FAM134B(family with sequence similarity 134,member B)等,均有高度保守的LIR序列,通过与LC3相互作用,促进多余的内质网组分或者受损的内质网转移至自噬体继而被降解,减轻ERS[86,87,88]。以上研究表明,多种自噬途径参与ERS的调控,自噬能够通过抑制ERS相关蛋白的表达或直接通过降解受损的内质网对ERS进行负反馈调节,避免细胞内应激水平加剧,维持胞内稳态,促进细胞存活,其中所涉及的信号通路以及ER-phagy中受损内质网如何与主体分离并转移到自噬体的机制尚不清晰,需要进一步深入研究。
4、 ERS-自噬参与人类疾病的发生发展
内质网作为蛋白质合成的场所,其稳态对维持蛋白质代谢平衡至关重要。内质网内未折叠或错误折叠蛋白的大量堆积会引起ERS。持续高水平的ERS与多种疾病的发生发展密切相关,如肾脏疾病、慢性肝损伤、帕金森病、糖尿病等。ERS诱导的自噬反应在清除内质网内疾病相关蛋白中起着相当重要的作用。先天性芬兰型肾病综合征由维持肾小球滤过屏障通透性的neptrin基因错义突变所致。Neptrin蛋白需要在内质网合成加工修饰后转移到细胞膜发挥作用,基因突变导致错误折叠的neptrin蛋白在内质网聚积,引起ERS,激活UPR,同时,neptrin蛋白的大量堆积引发细胞自噬,降解内质网中错误折叠的neptrin蛋白[89]。因此,增强细胞自噬可能会有效清除肾小球细胞中聚积的错误折叠蛋白,起到治疗肾病综合征的作用。α1-抗胰蛋白酶缺乏症会导致α1-抗胰蛋白酶Z型突变体在内质网中积聚,其与慢性肝损伤和肝癌的发生有关。α1-抗胰蛋白酶Z型突变体在内质网的积聚致使内质网膨胀,引起ERS,持续的应激会引发不依赖UPR及Ca2+的自噬途径清除积聚的蛋白[90]。帕金森病中标志性的α-synuclein异常折叠蛋白的出现以及黑质多巴胺能神经元的减少会导致ERS,持续的ERS会激活细胞自噬以清除错误折叠蛋白和损伤的内质网[91]。人类肌萎缩硬化由突变的dysferlin蛋白在内质网中积聚所致。在ATG5缺失的小鼠胚胎成纤维细胞中,dysferlin蛋白的聚积增加,诱发ERS,用雷帕霉素增强自噬后内质网中聚集的dysferlin蛋白得以减少甚至清除[92]。以上研究提示,可以用增强自噬的方法来治疗内质网突变蛋白异常积聚引起的疾病,而过度的自噬会导致细胞死亡,因此需要合理控制自噬的强度,使其适应不同组织细胞特定的生理或病理状态,达到抑制疾病发展,维持细胞正常功能的目的。如何对自噬进行适度的人为激活或抑制,有待于具体ERS相关疾病与自噬相互作用关系及其相关分子机制的充分阐明。
5、结语与展望
ERS和细胞自噬是细胞在程度不同的刺激因素下产生的适应性反应,二者相互作用,共同调节细胞命运。轻度的ERS引起的UPR是一种防御机制,维护细胞和机体的稳态。持续发生的ERS则能诱导细胞自噬,自噬发挥类似UPR的补充机制,进一步降解异常蛋白质和受损的内质网等细胞器,并通过负反馈机制抑制ERS,减弱环境刺激对细胞的影响,维持细胞稳态。通常情况下自噬对细胞起保护作用,但在一些强刺激因素下,自噬过度激活导致大量细胞质被降解,引起细胞自噬性死亡。例如,在引起ERS的低氧和高ROS条件下,适度的自噬能够缓解其对细胞的损害,促进细胞存活。但当严重缺氧和ROS持续升高时,自噬被过度激活,细胞则走向死亡。值得注意的是,自噬性细胞死亡是不同于细胞凋亡的细胞程序性死亡的另一种调控机制。在严重的ERS条件下,机体的适应性反应不足以消除ERS,也将引发细胞凋亡。此时一些自噬相关因子发挥促凋亡作用,如ATG蛋白和Beclin-1被凋亡蛋白质胱天蛋白酶剪切后,失去诱导自噬的能力,经过剪切的自噬蛋白质片段则获得了促凋亡功能[93,94]。ATG7通过改变溶酶体膜通透性促进溶酶体光损伤后的凋亡[95],自噬还可通过降解内源性凋亡抑制因子而发挥促凋亡作用。细胞自噬与凋亡在ERS中的转变是否存在某种标志性因子的改变,提示自噬相关因子作用的转变,目前尚无确切证据。关于ERS相关疾病中自噬保护与自噬性死亡,自噬与凋亡转变过程中具体节点和机制的阐明,将对我们理解相关疾病的病理和预后机制具有重要意义。近年来,随着对ERS诱导自噬调节通路的扩大研究,我们对ERS和细胞自噬的关系有了长足的认识,对ERS相关疾病的防治也因而有了一些应对策略,但有些关键问题尚未解决,比如:如何对自噬及ERS相关通路进行精确调控以避免或减少自噬从保护机制转为损伤机制?具体疾病中ERS与细胞自噬的相互作用及详尽机制是怎样的?自噬与凋亡参与ERS相关疾病的作用节点在哪?我们期待不久的将来,这些问题可以解决,在明确具体疾病ERS-自噬相互作用机制的基础上,通过对ERS-自噬的精确调节,为相关疾病的预防和治疗提供新的策略。
参考文献
[1]WANG M,KAUFMAN R J.Protein misfolding in the endoplasmic reticulum as a conduit to human disease[J].Nature,2016,529(7586):326-35.
[2]ALMANZA A,CARLESSO A,CHINTHA C,et al.Endoplasmic stress reticulum signalling-from basic mechanisms to clinical applications[J].FEBS J,2019,286(2):241-78.
[3]SMITH M,WILKINSON S.ER homeostasis and autophagy[J].Essays Biochem,2017,61(6):625-35.
[4]HETZ C,PAPA F R.The unfolded protein response and cell fate control[J].Mol Cell,2018,69(2):169-81.
[5]TAMEIRE F,VERGINADIS I I,KOUMENIS C.Cell intrinsic and extrinsic activators of the unfolded protein response in cancer:mechanisms and targets for therapy[J].Semin Cancer Biol,2015,33:3-15.
[6]WANG M,LAW M E,CASTELLANO R K,et al.The unfolded protein response as a target for anticancer therapeutics[J].Crit Rev Oncol Hematol,2018,127:66-79.
[7]ZHANG K,KAUFMAN R J.Signaling the unfolded protein response from the endoplasmic reticulum[J].J Biol Chem,2004,279(25):25935-8.
[8]SOU S N,ILIEVA K M,POLIZZI K M.Binding of human BiP to the ER stress transducers IRE1 and PERK requires ATP[J].Biochem Biophys Res Commun,2012,420(2):473-8.
[9]MOHAMED E,CAO Y,RODRIGUEZ P C.Endoplasmic reticulum stress regulates tumor growth and anti-tumor immunity:a promising opportunity for cancer immunotherapy[J].Cancer Immunol Immunother,2017,66(8):1069-78.
[10]MU Y P,OGAWA T,KAWADA N.Reversibility of fibrosis,inflammation,and endoplasmic reticulum stress in the liver of rats fed a methionine-choline-deficient diet[J].Lab Invest,2009,90(2):245-56.
[11]LEE A S.The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress[J].Methods,2005,35(4):373-81.
[12]MIZUSHIMA N.Autophagy:process and function[J].Genes Dev,2007,21(22):2861-73.
[13]SHIMIZU S.Biological roles of alternative autophagy[J].Mol Cells,2018,41(1):50-4.
[14]YU L,MCPHEE C K,ZHENG L,et al.Termination of autophagy and reformation of lysosomes regulated by mTOR[J].Nature,2010,465(7300):942-6.
[15]MIZUSHIMA N,LEVINE B.Autophagy in mammalian development and differentiation[J].Nat Cell Biol,2010,12(9):823-30.
[16]LEVINE B,KROEMER G.Autophagy in the pathogenesis of disease[J].Cell,2008,132(1):27-42.
[17]SANJUAN M A,DILLON C P,TAIT S W,et al.Toll-like receptor signaling in macrophages links the autophagy pathway to phagocytosis[J].Nature,2007,450(7173):1253-7.
[18]NAKAGAWA I.Streptococcus pyogenes escapes from autophagy[J].Cell Host Microbe,2013,14:604-6.
[19]WHITE E.The role for autophagy in cancer[J].J Clin Invest,2015,125:42-6.
[20]HARA T,NAKAMURA K,MATSUI M,et al.Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice[J].Nature,2006,441(7095):885-9.
[21]KIM J,KUNDU M,VIOLLET B,et al.AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1[J].Nat Cell Biol,2011,13:132-41.
[22]JUNG C H,JUN C B,RO S H,et al.ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery[J].Mol Biol Cell,2009,20:1992-2003.
[23]KUNDU M.ULK1,mammalian target of rapamycin,and mitochondria:linking nutrient availability and autophagy[J].Antioxid Redox Signal,2011,14(10):1953-8.
[24]RUSSELL R C,TIAN Y,YUAN H,et al.ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase[J].Nat Cell Biol,2013,15:741-50.
[25]FAN W,NASSIRI A,ZHONG Q.Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L)[J].Proc Natl Acad Sci USA,2011,108(19):7769-74.
[26]SIMONSEN A,TOOZE S A.Coordination of membrane events during autophagy by multiple class Pl3-kinase complexes[J].J Cell Biol,2009,186(6):773-82.
[27]ITAKURA E,MIZUSHIMA N.Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins[J].Autophagy,2010,6(6):764-76.
[28]NODA N N,INAGAKI F.Mechanisms of autophagy[J].Annu Rev Biophys,2015,44:101-22.
[29]POLSON H E,LARTIGUE J,RIGDEN D J,et al.Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation[J].Autophagy,2010,6(4):506-22.
[30]DOOLEY H C,RAZI M,POLSON H E,et al.WIPI2 links LC3 conjugation with PI3P,autophagosome formation,and pathogen clearance by recruiting Atg12-5-16L1[J].Mol Cell,2014,55(2):238-52.
[31]KAUFMANN A,BEIER V,FRANQUELIM H G,et al.Mole-cular mechanism of autophagic membrane-scaffold assembly and disassembly[J].Cell,2014,156(3):469-81.
[32]BAKULA D,MULLER A J,ZULEGER T,et al.WIPI3 and WIPI4β-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy[J].Nat Commun,2017,8:15637.
[33]MERCER T J,GUBAS A,TOOZE S A.A molecular perspective of mammalian autophagosome biogenesis[J].J Biol Chem,2018,293(15):5386-95.
[34]BANRETI A,SASS M,GRABA Y.The emerging role of acetylation in the regulation of autophagy[J].Autophagy,2013,9(6):819-29.
[35]LEE I H,FINKEL T.Regulation of autophagy by the p300 acetyltransferase[J].J Biol Chem,2009,284(10):6322-8.
[36]HUANG R,XU Y,WAN W,et al.Deacetylation of nuclear LC3 drives autophagy initiation under starvation[J].Mol Cell,2015,57(3):456-66.
[37]HUANG R,LIU W.Identifying an essential role of nuclear LC3 for autophagy[J].Autophagy,2015,11(5):852-3.
[38]WAN W,YOU Z,XU Y,et al.mTORC1 phosphorylates acetyltransferase p300 to regulate autophagy and lipogenesis[J].Mol Cell,2017,68(2):323-35.
[39]SU H,YANG F,WANG Q,et al.VPS34 acetylation controls its lipid kinase activity and the initiation of canonical and non-canonical autophagy[J].Mol Cell,2017,67(6):907-21.
[40]PARK J M,JUNG C H,SEO M,et al.The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14[J].Autophagy,2016,12(3):547-64.
[41]WAN W,YOU Z Y,ZHOU L,et al.mTORC1-regulated and HUWE1-mediated WIPI2 degradation controls autophagy flux[J].Mol Cell,2018,72(2):303-15.
[42] 夏丹.细胞自噬调控的研究进展[J].临床医学进展(XIA D.Advances in research on regulation of autophagy[J].Progress in Clinical Medicine),2019,9(3):163-79.
[43]MUNSON M J,GANLEY I G.MTOR,PIK3C3,and autophagy:signaling the beginning from the end[J].Autophagy,2015,11(12):2375-6.
[44]HORNUNG J P,KOPPEL H,CLARKE P G.Endocytosis and autophagy in dying neurons:an ultrastructural study in chick embryos[J].J Comp Neurol,1989,283(3):425-37.
[45]YORIMITSU T,NAIR U,YANG Z,et al.Endoplasmic reticulum stress triggers autophagy[J].J Biol Chem,2006,281(40):30299-304.
[46]PLACIDO A I,PEREIRA C M F,DUARTE A I,et al.The role of endoplasmic reticulum in amyloid precursor protein processing and trafficking:implications for Alzheimer’s disease[J].Biochim Biophys Acta,2014,1842(9):1444-53.
[47]LI J,NI M,LEE B,et al.The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells[J].Cell Death Differ,2008,15(9):1460-71.
[48]HETZ C.The unfolded protein response:controlling cell fate decisions under ER stress and beyond[J].Nat Rev Mol Cell Biol,2012,13(2):89-102.
[49]HARDING H P,ZHANG Y,BERTOLOTTI A,et al.Perk is essential for translational regulation and cell survival during the unfolded protein response[J].Mol Cell,2000,5(5):897-904.
[50]B’CHIR W,MAURIN A C,CARRARO V,et al.The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression[J].Nucleic Acids Res,2013,41(16):7683-99.
[51]KOUROKU Y,FUJITA E,TANIDA I,et al.ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion,an essential step for autophagy formation[J].Cell Death Differ,2007,14(2):230-9.
[52]WEIDBERG H,SHVETS E,ELAZAR Z.Biogenesis and cargo selectivity of autophagosomes[J].Annu Rev Biochem,2011,80:125-56.
[53]DING W X,NI H M,GAO W,et al.Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability[J].Am J Pathol,2007,171(2):513-24.
[54]ROUSCHOP K M,VAN D B T,DUBOIS L,et al.The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5[J].J Clin Invest,2010,120(1):127-41.
[55]RZYMSKI T,MILANI M,PIKE L,et al.Regulation of auto-phagy by ATF4 in response to severe hypoxia[J].Oncogene,2010,29(31):4424-35.
[56]TSAI D H,CHUNG C H,LEE K T.Antrodia cinnamomea induces autophagic cell death via the CHOP/TRB3/Akt/mTOR pathway in colorectal cancer cells[J].Sci Rep,2018,8(1):17424.
[57]NOVOA I,ZENG H,HARDING H P,et al.Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha[J].J Cell Biol,2001,153(5):1011-22.
[58]TAMAKI T,KAMATSUKA K,SATO T,et al.A novel trans-membrane protein defines the endoplasmic reticulum stress-induced cell death pathway[J].Biochem Biophys Res Commun,2017,486(1):149-55.
[59]WONG C H,ISKANDAR K B,YADAV S K.Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation[J].PLoS One,2010;5(4):e9996.
[60]OGATA M,HINO S,SAITO A,et al.Autophagy is activated for cell survival after endoplasmic reticulum stress[J].Mol Cell Biol,2006,26(24):9220-31.
[61]HOLLIEN J,WEISSMAN J S.Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response[J].Science,2006,313(5783):104-7.
[62]VIDAL R L,FIGUEROA A,COURT F A,et al.Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy[J].Hum Mol Genet,2012,21(10):2245-62.
[63]MRAGARITI A,LI H,CHEN T,et al.XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation[J].J Biol Chem,2013,288(2):859-72.
[64]HETZ C,CHEVET E,HARDING H P.Targeting the unfolded protein response in disease[J].Nat Rev Drug Discov,2013,12(9):703-19.
[65]CHEN X,ILIOPOULOS D,ZHANG Q,et al.XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway[J].Nature,2014,508(7494):103-7.
[66]FUJIMOTO T,YOSHIMATSU K,WATANABE K,et al.Overexpression of human X-box binding protein 1 (XBP-1) in colorectal adenomas and adenocarcinomas[J].Anticancer Res,2007,27:127-31.
[67]ROMERO-RAMIREZ L,CAO H,REGALADO M P.X box-binding protein 1 regulates angiogenesis in human pancreatic adenocarcinomas[J].Transl Oncol,2009,2(1):31-8.
[68]GOZUACIK D,KIMCHI A.Autophagy as a cell death and tumor suppressor mechanism[J].Oncogene,2004,23(16):2891-906.
[69]CAI Y,ARIKKATH J,YANG L,et al.Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders[J].Autophagy,2016,12(2):225-44.
[70]QIN L,WANG Z,TAO L,et al.ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy[J].Autophagy,2010,6(2):239-47.
[71]WANG J,KANG R,HUANG H,et al.Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression[J].Autophagy,2014,10(5):766-84.
[72]RUTKOWSKI D T,ARNOLD S M,MILLER C N,et al.Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins[J].PLoS Biol,2006,4(11):e374.
[73]LIN J H,LI H,YASUMURA D,et al.IRE1 signaling affects cell fate during the unfolded protein response[J].Science,2007,318(5852):944-9.
[74]AISA Z,LIAO G C,SHEN X L,et al.Effect of autophagy on myocardial infarction and its mechanism[J].Eur Rev Med Pharmacol Sci,2017,21(16):3705-13.
[75]KOIKE M,SHIBATA M,TADAKOSHI M,et al.Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury[J].Am J Pathol,2008,172:454-69.
[76]ZHOU Y,SHU F R,LIANG X Y,et al.Ampelopsin induces cell growth inhibition and apoptosis in breast cancer cells through ROS generation and endoplasmic reticulum stress pathway[J].PLoS One,2014,9(2):e89021.
[77]KIM M J,WOO S J,YOON C H,et al.Involvement of auto-phagy in oncogenic K-Ras-induced malignant cell transformation[J].J Biol Chem,2011,286(15):12924-32.
[78]DADAKHUJAEV S,JUNG E J,NOH H S,et al.Interplay between autophagy and apoptosis in TrkA-induced cell death[J].Autophagy,2009,5(1):103-5.
[79]HOYER-HANSEN M,BASTHOLM L,SZYNIAROWSKI P,et al.Control of macroautophagy by calcium,calmodulin-dependent kinase kinase-beta,and Bcl-2[J].Mol Cell,2007,25(2):193-205.
[80]ZALCKVAR E,BERISSI H,EISENSTEIN M,et al.Phos-phorylation of Beclin1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL[J].Autophagy,2009,5(5):720-2.
[81]GROTEMEIER A,ALERS S,PFISTERER S G,et al.AMPK-independent induction of autophagy by cytosolic Ca2+ increase[J].Cell Signal,2010,22(6):914-25.
[82]SAKAKI K,KAUFMAN R J.Regulation of ER stress-induced macroautophagy by protein kinase C[J].Autophagy,2008,4(6):841-3.
[83]LI X,ZHU G,GOU X,et al.Negative feedback loop of autophagy and endoplasmic reticulum stress in rapamycin protection against renal ischemia-reperfusion injury during initial reperfusion phase[J].FASEB J,2018,doi:10.1096/fj.201800299R.
[84]MICHALAK M,ROBERT P J M,OPAS M.Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum[J].Cell Calcium,2002,32(5/6):269-78.
[85]YANG Y,MA F,LIU Z,et al.The ER-localized Ca2+-binding protein calreticulin couples ER stress to autophagy by associating with microtubule-associated protein 1A/1B light chain 3[J].J Biol Chem,2019,294(3):772-82.
[86]SONG S,TAN J,MIAO Y,et al.Crosstalk of ER stress-mediated autophagy and ER-phagy:involvement of UPR and the core autophagy machinery[J].J Cell Physiol,2018,233(5):3867-74.
[87]FUMAGALLI F,NOACK J,BERGMANN T J,et al.Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery[J].Nat Cell Biol,2016,18(11):1173-84.
[88]KHAMINETS A,HEINRICH T,MARI M,et al.Regulation of endoplasmic reticulum turnover by selective autophagy[J].Nature,2015,522(7556):354-8.
[89]李赫宁,李兰芳,陈临溪.内质网自噬:疾病防治的新靶标[J].中国药理学通报(LI H N,LI L F,CHEN L X.Endoplasmic reticulum autophagy:a new target for disease prevention[J].Chinese Pharmacological Bulletin),2015,31(3):302-8.
[90]KAMIMOTO T,SHOJI S,HIDVEGI T,et al.Intracellular inclusions containing mutant alphal-antitrypsin Z are propagated in the absence of autophagic activity[J].J Biol Chem,2006,281(7):4467-76
[91]GHAVAMI S,SHOJAEI S,YEGANEH B,et al.Autophagy and apoptosis dysfunction in neurodegenerative disorders[J].Prog Neurobiol,2014,112:24-49.
[92]FUJITA E,KOUROKU Y,ISOAI A,et al.Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin:ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II)[J].Hum Mol Genet,2007,16(6):618-29.
[93]SHI M,ZHANG T,SUN L,et al.Calpain,Atg5 and Bak play important roles in the crosstalk between apoptosis and autophagy induced by influx of extracellular calcium[J].Apoptosis,2013,18(4):435-51.
[94]HUANG X,QI Q,HUA X,et al.Beclin1,an autophagy-related gene,augments apoptosis in U87 glioblastoma cells[J].Oncol Rep,2014,31(4):1761-7.
[95]KESSEL D H,PRICE M,REINERS J J.ATG7 deficiency suppresses apoptosis and cell death induced by lysosomal photodamage[J].Autophagy,2012,8(9):1333-41.

1963年,Duve首次提出自噬这一概念,该现象是基于观察到经胰高血糖素诱导的小鼠肝细胞内降解的线粒体及其它的内在结构。近年来,医学界重新认识自噬,在分子水平上一定程度地了解了这个过程的重要意义。目前,人们在酵母菌内已经发现了32个不同种类的自噬相关基因...
自噬是由细胞质起源的自噬体与溶酶体融合降解胞内异常分子和长寿命蛋白的过程,是一种特殊分解代谢途径。它在细胞代谢、存活与分化等方面都具有重要作用。当前社会正朝向老龄化社会演变,关注老龄化相关疾病的研究显得格外重要。随着细胞生物科学与老年医学...
自噬(autophagy)是由Ashford和Porter[1]在1962年发现细胞内存在自己吃自己的现象后提出,系指由细胞粗面内质网或高尔基体等脱落的双层膜包裹部分细胞质和受损的细胞器及蛋白质,而后与溶酶体融合降解其所包裹内容物,从而实现细胞本身的代谢需要和细胞...
自噬是广泛存在于真核生物中的保守的代谢过程,是细胞成分更新、发育、分化的重要调控机制,对维持蛋白代谢平衡及细胞内环境稳定具有重要意义。转化生长因子(Transforminggrowthfactor,TGF-)是一种具有多种功能的多肽生长因子,可促进细胞的增殖与分化...
自噬(autophagy)是真核细胞利用溶酶体降解损伤的长寿蛋白及细胞器的过程,通过对细胞降解成分的再利用在维持内环境稳态中起着非常重要的作用。正常生理情况下,它维持在较低的水平,清除多余、受损的细胞器,为细胞提供保护作用。在病理情况下,氧化应激...
0引言内质网应激(endoplasmicreticulumstress,ERS)是指在多种生理或病理条件下细胞内质网钙稳态失衡或蛋白质加工运输障碍、生理功能发生紊乱的一种亚细胞器的病理过程[1-3],涉及内质网类似激酶(proteinkinaseRNA-likeendoplas-micreticulumkinase...
炎症体的概念最早由Martinon等[1]提出,其来源主要是模式识别受体(patternrecognitionreceptors,PRRs)中的核苷酸结合寡聚化结构域(nucleotide-bindingoligomerizationdomain,NOD)样受体(NOD-likereceptors,NLRs)在活化含半胱氨酸的天...
近年来,越来越多的学者发现自噬与人体多种纤维化疾病有关,细胞自噬可以减轻纤维化的发展,但过度的自噬会加重纤维化疾病的发生,所以这需要广大的研究者进一步研究自噬的发生机制和发展规律,从而使自噬成为我们治疗纤维化疾病新的治疗方式。...