文献求助

求助主题Targeting FTO Suppresses Cancer Stem Cell Maintenance and Im
需求说明求助文献
求助时间2021-05-30 12:22
您好!学术堂根据您的需求,查找到英文名为《Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion》的外文文献1篇,以下是文献部分内容展示(不含图表),本页末尾提供完整全文pdf免费下载。
Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion
SUMMARY
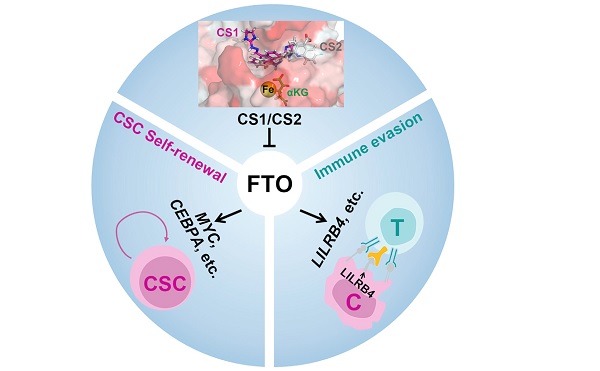
Fat mass and obesity-associated protein (FTO), an RNA N6-methyladenosine (m6A) demethylase, playsoncogenic roles in various cancers, presenting an opportunity for the development of effective targeted ther-apeutics. Here, we report two potent small-molecule FTO inhibitors that exhibit strong anti-tumor effects inmultiple types of cancers. We show that genetic depletion and pharmacological inhibition of FTO dramati-cally attenuate leukemia stem/initiating cell self-renewal and reprogram immune response by suppressingexpression of immune checkpoint genes, especially LILRB4. FTO inhibition sensitizes leukemia cells toT cell cytotoxicity and overcomes hypomethylating agent-induced immune evasion. Our study demonstratesthat FTO plays critical roles in cancer stem cell self-renewal and immune evasion and highlights the broadpotential of targeting FTO for cancer therapy.
INTRODUCTION
Among the >170 modified RNA nucleotides, N6-methyladeno-sine (m6A) represents the most abundant and prevalent internalmodification in eukaryotic mRNA (Boccaletto et al., 2018; Fryeet al., 2018). Fat mass and obesity-associated protein (FTO)was identified as the first RNA demethylase that canremove m6A from RNA through an a-ketoglutarate (a-KG) and Fe(II)-dependent mechanism (Jia et al., 2011), suggesting thatm6A is a type of reversible and dynamic RNA modification (Jiaet al., 2013). Recently, we reported that FTO is overexpressedand plays a critical role in leukemia as an m6A demethylase (Liet al., 2017). Subsequently, we showed that FTO is a target ofR-2-hydroxyglutarate (R-2HG) and by suppression of FTO activ-ity, R-2HG displays intrinsic anti-leukemia effects (Su et al.,2018). In addition, the aberrant overexpression and potentialoncogenic roles of FTO have also been reported in multiple solidtumors (Niu et al., 2019; Tang et al., 2019; Yang et al., 2019).
Thus, these data suggest that FTO is a promising therapeutictarget. Better understanding of the mechanisms underlyingFTO’s functions in cancers and development of effective tar-geted therapeutics against FTO are warranted.
A set of specific or non-specific FTO inhibitors, such as rhein,meclofenamic acid (MA), MO-I-500, fluorescein, and R-2HG,have been identified (Chen et al., 2012; He et al., 2015; Huanget al., 2015; Padariya and Kalathiya, 2016; Singh et al., 2016;Su et al., 2018; Toh et al., 2015; Wang et al., 2015; Zhenget al., 2014). However, all these small molecules are limited inclinical potential due to mild biological function and low sensi-tivity and/or specificity (Huang et al., 2019). More recently, twoderivatives of MA, termed FB23 and FB23-2, have been devel-oped, which showed improved efficacy in inhibiting FTO activityand viability of human acute myeloid leukemia (AML) cells. None-theless, their IC50values in inhibiting AML cell viability are still>1 mM (FB23-2) or even >20 mM (FB23) (Huang et al., 2019).
While FB23-2 showed a statistically significant effect on inhibit-ing the progression of human primary AML in mice, which pro-vides proof-of-concept evidence indicating the therapeutic po-tential of pharmacological targeting FTO in treating AML, theinhibitory degree was not satisfactory. Thus, there is still an ur-gent and unmet need to develop efficacious inhibitors againstFTO to treat AML and other cancers.
Here, through a series of screening and validation assays, weidentified two potent small-molecule FTO inhibitors. Our furtherstudies revealed the significant effects and the underlying mech-anisms of targeting FTO on suppressing cancer stem cell self-renewal and immune evasion, highlighting the broad potentialof targeting FTO for cancer therapy.
RESULTS
Identification of Effective FTO Inhibitors
To identify potential FTO inhibitors, we conducted a structure-based virtual screening of the 260,000 compounds from the Na-tional Cancer Institute Developmental Therapeutics Program(NCI DTP) library (see STAR Methods for details). We requestedfrom NCI the top 370 candidate compounds that showed thehighest scores based on their docking to FTO’s catalytic pocket(Figures 1A-1C), but only 213 compounds were available. Wethen assessed their anti-leukemic efficacy in the humanMONOMAC 6 AML cell line (carrying t(9; 11)/MLL-AF9) via MTT(3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide)cell proliferation/viability assays (Figure S1A). The top 20 com-pounds showing the most robust inhibitory effects on MONO-MAC 6 cell viability (Figure 1D) were selected and furthervalidated in two additional AML cell lines (NOMO-1 and U937;Figure S1B). We also assessed their efficacy on inhibition ofFTO’s m6A demethylase activity through cell-free m6A demethy-lase assays (Figure 1E). We identified three compounds (CS1,CS2, and NSC 48890) that display consistently robust effectson inhibition of AML cell viability and FTO’s demethylase activity.
Due to the overly simplistic structure of NSC 48890 (unlikely a se-lective inhibitor) (Figure S1C), we decided to focus on CS1 andCS2 (Figure 1F) for further studies.
Our docking models suggest that both CS1 and CS2 bindtightly to FTO protein and block its catalytic pocket (Figures1G-1J, S1D, and S1E). Additionally, based on the crystal struc-ture of FTO-oligonucleotide complex (Zhang et al., 2019), wefound that CS1/2 interact with FTO residues that were knownto be involved in the binding of FTO with m6A modified single-strand DNA (Zhang et al., 2019), such as HIS231 and GLU234by CS1, and LYS216, SER229, and HIS231 by CS2 (Figures1H-1J and S1F-S1H). These data suggest that CS1 and CS2selectively bind to and occupy the catalytic pocket of FTO andthereby block m6A-modified oligos from entering into FTO’s cat-alytic pocket, which in turn inhibits FTO’s demethylase activityand its binding with the target RNA transcripts.
CS1 and CS2 Are Highly Efficacious FTO Inhibitors withPotent Anti-leukemic Efficacy In Vitro
Compared with two previously reported FTO inhibitors (FB23-2and MO-I-500) (Huang et al., 2019; Zheng et al., 2014), CS1and CS2 displayed a much higher efficacy in inhibiting AMLcell viability, with 10- to 30-fold lower IC50(half-maximal inhibi-tory concentration) values in AML cells (Figures S2A and S2B),indicating their greatly improved efficacy. We then determinedtheir IC50values in a panel of leukemia cell lines with high orlow levels of FTO expression (Li et al., 2017; Su et al., 2018).
As expected, the FTO-high leukemia lines showed lower IC50values than the FTO-low cell lines (Figures 2A and 2B). Knock-down (KD) of FTO in FTO-high AML cells reduced their sensitivityto CS1 and CS2 (Figures S2C and S2D). These results suggestthat the anti-leukemia effects of CS1 and CS2 are FTO-abun-dance dependent. Both CS1 and CS2 significantly inhibited theviability of human primary AML cells, but largely spared thehealthy control cells (Figures 2C and 2D), highlighting their ther-apeutic potential in treating leukemia patients.
The direct interactions between CS1/2 and FTO protein wereconfirmed by a biophysical method, nuclear magnetic reso-nance (NMR). CS1 and CS2-induced dose-dependent attenua-tion of signals were observed in Carr-Purcell-Meiboom-Gill(CPMG) NMR titration, and positive saturation transfer signals(STD) were also detected (Figures 2E-2H), demonstrating theirdirect binding with FTO in vitro (i.e., in a cell-free system). Drug affinity responsive target stability (DARTS) (Lomenick et al.,2009) assay and cellular thermal shift assay (CETSA) (Jafariet al., 2014) were conducted to confirm their direct interactionsin AML cells. According to the docking poses of CS1/2 andFTO protein (see Figures 1I and 1J), residues H231 and E234are essential for the binding of FTO with CS1, while K216,S229, and H231 are crucial for its binding with CS2. CS1 andCS2 could block pronase-induced proteolysis of wild-type(WT) FTO but not that of mutant FTOH231A/E234Aor FTOK216A/S229A/H231A(Figures 2I-2L). Such data confirmed that FTO binddirectly with CS1 and CS2 in cellulo, and the mutated aminoacids are essential for their interactions. In addition, both CS1and CS2 treatment led to substantial shifts of the thermal stabilityof FTO protein (Figure 2M), which further confirmed their directinteractions. Through cell-free m6A demethylase assays, weshowed that both CS1 and CS2 efficiently suppressed m6A de-methylase activity of FTO, with IC50values in the nanomolarrange (Figures 2N and S2E).
Since the residues K216, S229, H231, and E234 of FTO areessential for the bindings of FTO with both CS1/2 (Figures 2I-2L) and m6A-modified oligonucleotides (Zhang et al., 2019), wepresumed that CS1 and CS2 could disrupt the binding of FTOwith its target RNAs. Indeed, our crosslinking immunoprecipita-tion-qPCR data confirmed that CS1 and CS2 block the bindingof FTO with its known target mRNAs, such as MYC, CEBPA,and RARA (Li et al., 2017; Su et al., 2018) (Figures S2F-S2H).
In addition, CS1 and CS2 treatment notably increased globalm6A abundance in AML cells (Figure S2I) but had no noticeableeffect on the FTO protein level (Figure S2J). Neither CS1 nor CS2treatment suppressed the enzymatic activity of ALKBH5,another major m6A demethylase (Zheng et al., 2013), or TET1,another a-KG-dependent dioxygenase (Figures S2K and S2L),highlighting the selectivity of CS1 and CS2 against FTO.
Effects of FTO KD and Inhibition on AML Cell Viabilityand Differentiation and on Leukemia Stem/InitiatingCell Self-Renewal
Consistent with the effects of FTO KD (Li et al., 2017), we showedthat pharmacological inhibition of FTO by CS1 or CS2 resulted insubstantially increased apoptosis and cell-cycle arrest (at the G0phase) in human AML cells (Figures 3A-3D and S3A-S3D). Bothinhibitors, alone or together with all-trans retinoic acid, alsosignificantly promoted myeloid differentiation in human AMLcells (Figures S3E and S3F).
Leukemia stem/initiating cells (LSCs/LICs), characterized bytheir unlimited self-renewal potential, are considered to be theroot cause of the treatment failure and relapse of AML; thus,eradication of LSCs/LICs is necessary to achieve a cure (Krauseand Van Etten, 2007; Pollyea and Jordan, 2017). Via flow cytom-etry, we found that FTO is overexpressed in human primary AMLpatient cells relative to healthy control cells (Figures 3E andS3G). Moreover, in primary AML patient samples, the FTO levelis even higher in CD34+immature AML cells than in CD34AML bulk cells (Figures 3F and 3G). Consistent with their higherFTO levels, AML patient samples have a lower m6A abundancecompared with healthy controls (Figure S3H), as do CD34+AML cells compared with CD34 AML cells (Figure S3I). FTOKD substantially promoted apoptosis and myeloid differentiationand suppressed the colony-forming capability of human primaryAML CD34+ cells (Figures S3J-S3L), implying that FTO may playa role in self-renewal/repopulation of LSCs/LICs. To test this, weconducted in vitro and in vivo limiting dilution assays (Krivtsovet al., 2006; Li et al., 2015; Weng et al., 2018). Either KD ofFTO or pharmacological inhibition of FTO resulted in a remark-able decrease in the frequency of LSCs/LICs in murine AMLmodels (Figures 3H-3L). Notably, 50 nM CS1 could almostcompletely inhibit the repopulating capacity of AML cells (Fig-ure S3M), further highlighting the potent effect of our FTO inhib-itors in suppressing self-renewal of LSCs/LICs.
CS1 and CS2 Treatments Modulate the SignalingPathways of FTO
RNA sequencing (RNA-seq) was carried out to understand themolecular mechanisms underlying the effects of CS1 and CS2(Figure S4A). Cluster analysis revealed that CS1, shFTO, andCS2 treated samples can be grouped together, separate fromthe two control groups (Figure 4A). The dysregulated genesinduced by CS1, CS2, and FTO KD overlapped well with eachother (Figures 4B and 4C). Our RNA-seq and qPCR data showedthat CS1 or CS2 treatment substantially decreased MYC andCEBPA expression while increasing RARA and ASB2 expression(Figures 4D, S4B, and S4C), which are positive and negativetargets of FTO, respectively (Li et al., 2017; Su et al., 2018). Bytargeting FTO, CS1 and CS2 treatment also increased m6Aabundance on FTO target RNAs, such as MYC and CEBPAmRNA (Su et al., 2018) and small nuclear RNAs (snRNAs) (Maueret al., 2019) (Figures S4D-S4F). Through global gene set enrich-ment analysis (GSEA) (Subramanian et al., 2005), we identified aset of upregulated or downregulated pathways upon CS1 or CS2treatment or FTO KD. Notably, among the upregulated path-ways, CS1, CS2, and shFTO groups shared the majority of theirenriched signaling pathways and core-enriched genes (Figures4E [left panel] and 4F; Table S2). Among the downregulatedpathways, all the pathways suppressed by CS1 or CS2 also existin the pathways suppressed by FTO KD (Figures 4E [right panel]
and 4G; Table S2). FTO inhibition or KD-mediated cell apoptosis and cell-cycle arrest are likely attributed to the activation of the‘‘Apoptosis’’ signaling, and suppression of the ‘‘MYC targetsV1’’ and ‘‘MYC targets V2’’ pathways (Figure 4H). We alsocompared the key biological pathways effects by CS1, CS2,and other FTO inhibitors, FB23 and FB23-2 (Huang et al.,2019), and found that the distinct inhibitors shared these crucialsignaling pathways and core-enriched gene (Figure S4G and Ta-ble S2). Thus, our mechanistic study data suggest that CS1 andCS2 exert their anti-leukemic effects through modulation of theessential signaling pathways of FTO.
CS1 and CS2 Display Potent Anti-leukemic Efficacy In Vivo
We next assessed the therapeutic efficacy of CS1 and CS2in vivo. In a patient-derived xenotransplantation (PDX) AMLmodel (with a relapsed AML patient sample, 2017-38), weshowed that CS2 treatment dramatically reduced leukemiainfiltration (Figure S5A) and doubled the overall survival (Fig-ure 5A). Surprisingly, however, CS1 treatment did not showany significant effects, although CS1 exhibited an equal oreven stronger anti-leukemic activity compared with CS2in vitro (see Figures 2A, 2B, and 3A-3D). Further analysis re-vealed that the poor solubility and uptake of CS1 likely causedits weak effect in vivo. To increase bioavailability, we employedmPEG-b-PLA micelles or b-cyclodextrin, both widely used inthe clinic (Cho et al., 2016; Hirayama and Uekama, 1999), todeliver hydrophobic CS1 (Figure 5B). We then repeated thetreatment with the same PDX AML model by use of micellespackaged CS1 (Micelle_CS1), and demonstrated that deliveryof CS1 with micelles markedly improved its anti-leukemia ac-tivity in vivo (Figure 5C). Similarly, Micelle_CS1 displayed amuch more potent anti-leukemic activity than free CS1 in treat-ing mice bearing transplanted murine MLL-AF9 AML, wherefree CS2 still showed robust anti-leukemic activity (Figures5D, S5B, and S5C). Both Micelle_CS1 and free CS2 also dis-played potent anti-AML efficacy in another PDX AML model(AML 3448), significantly more effective than FB23-2(Figures S5D-S5F). Similarly, b-cyclodextrin packaged CS1(b-CD_CS1) and CS2 substantially delayed AML progressionand prolonged survival in an additional PDX model (AML,2016-9) (Figures 5E, 5F, and S5G) accompanied by a signifi-cant impact on the expression of FTO targets, includingMYC, RARA, and ASB2 (Figure S5H). Via bioluminescence im-aging, we observed that pharmacological inhibition or KD ofFTO remarkably inhibited leukemia progression, constantlyreduced leukemia burden, and dramatically prolonged survival(Figures 5G, 5H, and S5I-S5K). Thus, our preclinical animalmodel studies demonstrated the potent therapeutic efficacyof CS1 (packaged by micelles or b-cyclodextrin) or CS2 alonein treating AML, including relapsed AML. As we just testedrelatively low dosages of CS1 and CS2 (merely 5 mg/kgonce every other day, 10 times), higher dosages may resultin more robust therapeutic effects.
The FTO/m6 A Axis Regulates Immune Checkpoint Gene Expression
We previously reported that R-2HG-mediated FTO inhibition dis-played synergistic effects with hypomethylating agents (HMAs;e.g., azacitidine and decitabine [DAC]) in treating AML (Suet al., 2018). HMAs are wildly used for the treatment of patientswith AML or myelodysplastic syndrome (MDS), especially inelderly patients and in those who are not eligible for allogeneicstem cell transplantation (Dombret et al., 2015; Issa et al.,2004; Yun et al., 2016). However, the vast majority of AML orMDS patients treated with HMA eventually developed drug resis-tance (Yun et al., 2016). The upregulation of immune checkpointgenes, such as PD-1, PD-L1, and PD-L2, and subsequent im-mune evasion have been assumed to contribute to HMA-induced drug resistance in the treated patients with myeloid ma-lignancies (Orskov et al., 2015; Stahl and Goldberg, 2019; Yanget al., 2014). Since inhibition of FTO by R-2HG could sensitizeAML cells to HMA (Su et al., 2018), here we sought to revealthe mechanism(s) underlying their synergistic effect and deter-mine whether FTO signaling contributes to HMA-mediated upre-gulation of immune checkpoint genes and subsequent immuneevasion.
We confirmed the increased expression of PD-L1, PD-L2, andPD-1 upon DAC treatment in human AML or T cells (Figure S6A).
Strikingly, DAC treatment also resulted in globally decreasedm6A abundance in AML cells (Figure 6A). The reduced m6A levelis likely the result of the increased expression of m6A eraser FTO,as no significant expression changes observed in ALKBH5,METTL3, or METTL14 (Figures 6B and S6B). We thus presumedthat DAC-induced FTO overexpression may contribute to theincreased expression of immune checkpoint genes via an m6A-dependent mechanism. Indeed, FTO KD or inhibition signifi-cantly inhibited the expression of PD-L1 and PD-L2 in humanAML cells with or without DAC treatment (Figures S6C-S6F).
Nonetheless, we found that the endogenous expression levelsof such immune checkpoint genes in most human AML cell linesare very low (Figures S6G-S6I). Consistently, it was reported thatdue to their limited expression in AML patients, targeting thoseimmune checkpoint proteins by inhibitors alone showed onlylimited clinical efficacy in treating AML patients (Berger et al.,2008; Daver et al., 2019).
Besides PD-L1/PD-L2, leukocyte immunoglobulin-like recep-tor subfamily B (LILRB), including LILRB1, 2, 3, 4, and 5, havealso been recognized as immune checkpoint proteins in AML(Chen et al., 2018; Deng et al., 2018a; Kang et al., 2016). Weobserved that DAC treatment remarkably promoted expression of LILRB family members, including LILRB3, LILRB4, andLILRB5 (Figure S6J). In particular, the LILRB4 level wasincreased over 100-fold upon DAC treatment (Figure 6C), whichis 6= to 20-fold greater than the fold changes for PD-L1 and PD-L2 (see Figure S6A). Interestingly, among the LILRB genes, onlyexpression of LILRB4 could be significantly downregulated byFTO KD (Figure S6K), implying that LILRB4 might be a target ofFTO in AML cells. Notably, LILRB4 is overexpressed in humanAML cell lines relative to normal bone marrow-derived mononu-clear cells (MNCs) and T cells (Figures 6D and S6L), with a muchhigher level than those of PD-1, PD-L1, and PD-L2 in AML lines(Figure S6M). Similarly, when analyzing The Cancer GenomeAtlas (TCGA) AML dataset (Ley et al., 2013), we found that themedian expression level of LILRB4 is 40- to 50-fold higher thanthose of PD-L1 and PD-L2 in human primary AML samples(Figure 6E).
Consistent with the effect of FTO KD, CS1 or CS2 treatmentalso significantly decreased LILRB4 expression at both RNAand protein levels; conversely, forced expression of FTO WT(but not the catalytically inactive mutant) significantly increasedthe expression of LILRB4 (Figures 6F-6L). DAC treatment couldpartially rescue the suppressed expression of LILRB4 inducedby FTO inhibition (Figure 6M). Notably, CS1 and CS2 treatmentdid not obviously affect LILRB4 level in normal dendritic cellsor macrophages (Figures S6N and S6O), likely due to the lowlevel of FTO in such cells. FTO inhibition or KD increased m6Aabundance on LILRB4 mRNA transcript (Figures 6N and 6O).
Moreover, we demonstrated that FTO WT, but not mutant, couldsignificantly increase the stability of LILRB4 mRNA in AML cells;the opposite is true when FTO was knocked down (Figures 6Pand 6Q). KD of m6A reader YTHDF2, which was reported to pro-mote decay of m6A-modified transcripts (Wang et al., 2014), alsoincreased the half-life of LILRB4 mRNA (Figure 6R). Together,the results show that FTO positively regulates LILRB4expression in AML by suppressing YTHDF2-mediated decay ofm6A-modified LILRB4 mRNA.
Targeting FTO Sensitizes AML Cells to T Cell Cytotoxicity and Overcomes HMA-Induced Immune Evasion
To determine whether pharmacological inhibition of the FTO/m6A/LILRB4 axis can reprogram immune response, we pre-treated AML cells with CS1 or CS2 and then co-cultured themwith activated T cells. We found that FTO inhibition sensitizedhuman AML cells to T cells, accompanied by decreased expres-sion of LILRB4 in AML cells (Figures 7A-7E). To generate acomprehensive molecular profiling of FTO inhibition in an im-mune-competent setting, we utilized the MLL-AF9 AML mousemodel for RNA-seq. Among the immune checkpoint genes,Lilrb4 is highly expressed in AML cells and significantly sup-pressed by FTO inhibitor therapy (Figures 7F, 7G, and S7A).
Further flow-cytometry studies validated the downregulation ofLilrb4 by CS1 and CS2 treatment in vivo (Figures 7H-7K). LILRB4knockout (KO) or KD also significantly inhibited human AML cellgrowth (Figures 7L and 7M). Consistent with its role in immunesurveillance, forced expression of LILRB4 suppressed T cellkilling of human AML cells with or without FTO inhibitor pretreat-ment (Figures 7N, 7O, S7B, and S7C).
To assess the effect of targeting the FTO/m6A/LILRB4 axis onAML progression and immune evasion in vivo, we employedAML xenograft models with FTO inhibition and T cell treatment.
We found that FTO inhibition (by CS1 or CS2) synergized withT cell treatment and substantially suppressed AML progression,resulting in remarkably prolonged survival in the combinationaltreatment groups (Figures 8A and 8B). Consistent with the roleof FTO in mediating HMA-induced upregulation of immunecheckpoint genes and subsequent immune evasion, FTO inhibi-tion also synergized with HMAs (e.g., DAC) in inhibiting AMLprogression in immune-competent BMT recipient mice, andthe combinations showed much improved therapeutic efficacythan either treatment alone (Figures 8C, S7D, and S7E). Collec-tively, FTO inhibition could suppress immune checkpoint geneexpression and thereby sensitize AML cells to T cell cytotoxicityand overcome HMA-induced immune evasion.
The Minimal Drug Toxicity, Structure-Activity Relationships, and Broad Anti-cancer Efficacy of FTO Inhibitors
To evaluate the potential drug toxicity of CS1 and CS2 in vivo, weinjected two doses for each compound (5 mg/kg/day [i.e., thedose used for AML mouse treatment] and 20 mg/kg/day) intoC57BL/6 mice once every other day for 20 days, and euthanizedall the mice 10 days after the final treatment. We observed no sig-nificant difference between the drug-treated groups and controlgroup regarding whole-body or organ weight (Figures S7F-S7Kand Table S3). Complete blood count data collected from periph-eral blood did not show any significant difference between thetreated groups and control group (Table S3). H&E staining alsoshowed no difference between the groups (Figure S7L). Thesedata suggest that the drug toxicity of CS1 or CS2 is minimal.
To explore the structure-activity relationships of CS1 and CS2chemical scaffolds, we designed and synthesized six analogcompounds for CS1 and four analogs for CS2, based on theirstructures and their binding poses with FTO protein. Amongthe six CS1 analogs, only CS1-3 and CS1-7 showed anti-leukemic efficacy similar to that of CS1 (Figure S8A). As shownin the docking models, both CS1-3 and CS1-7 display tight bind-ing with FTO protein (Figure S8A). According to the structures ofCS1 and its analogs, we conjecture that the planar three-ringstructure may be important for their efficacy. Among CS2 ana-logs, only CS2-2, with the highest similarity to CS2, exhibitedan anti-leukemia effect similar to that of CS2 (Figure S8B).
Further systematic studies are warranted to develop more effec-tive CS1 and CS2 analogs.
In addition to hematopoietic malignancies, FTO has also beenreported to play oncogenic roles in many types of solid tumors(Huang et al., 2020a; Niu et al., 2019; Tang et al., 2019; Yanget al., 2019). To test the therapeutic potential of targeting FTOin treating solid tumors, we chose glioblastoma, breast cancer,and pancreatic cancer as representative models, in which FTOexpression is comparable with that in AML (Figure S8C). FTOKD or inhibition significantly suppressed the proliferation ofthese cancer cells (Figures S8D-S8G). Further in vivo studiesconfirmed the potent anti-tumor efficacy of FTO inhibitors intreating breast cancer (Figures 8D and 8E). Together, our resultsdemonstrate the broad therapeutic potential of FTO inhibitors intreating various types of cancers.
DISCUSSION
While m6A modification and the machinery have been implicatedin the initiation, progression, maintenance, and drug resistanceof various types of cancers (Deng et al., 2018b, 2018c; Huanget al., 2020b), the development of effective inhibitors to targetm6A regulators for cancer therapy is still in its infancy (Huanget al., 2020a). In the present study, by in silico virtual screeningand subsequent validation assays, we have identified two effec-tive small-molecule compounds (CS1 and CS2) that specificallytarget FTO and efficiently suppress its m6A demethylase activityby occupying the catalytic pocket and interfering with the bind-ing of FTO with m6A-modified RNAs. CS1 and CS2 treatmentsignificantly inhibited the viability/growth of human AML cellswith IC50values at low nanomolar levels, which are at least 10-fold more effective than previously reported FTO inhibitors(e.g., FB23-2 and MO-I-500) (Huang et al., 2019; Zheng et al.,2014). Mechanistically, CS1 and CS2 exert anti-leukemic effectsby suppression of FTO activity and signaling, leading to the acti-vation of apoptosis signaling and inhibition of MYC pathways.
Notably, CS1 (NSC337766, also named bisantrene) has beenintroduced into clinical trials since the 1980s as an anthracenecompound for various types of cancer therapy, and some pa-tients responded to such treatment (Cowan et al., 1986; Milleret al., 1986; Myers et al., 1984; Pratt et al., 1986; Rothman,2017; Yap et al., 1983). This agent was originally thought to besimilar to doxorubicin in activity (Yap et al., 1983); however, un-like doxorubicin, bisantrene does not exhibit anthracycline-associated cardiotoxicity and was generally well tolerated bymost patients (Rothman, 2017; Yap et al., 1983). Indeed, asidefrom functioning as a DNA-reactive agent, its immune-activatingand telomerase-inhibiting activities have also been reported(Rothman, 2017), suggesting that the mechanisms of its actionhave yet to be fully investigated. CS2 (NSC368390, also namedbrequinar) was previously reported to inhibit the enzyme dihy-droorotate dehydrogenase and thereby block de novo pyrimi-dine biosynthesis (Peters et al., 1992). Brequinar has also beentested in clinical trials for cancer therapy (Burris et al., 1998; deForni et al., 1993; Noe et al., 1990; Schwartsmann et al., 1990).
In the present study, we demonstrated that CS1 and CS2 binddirectly to FTO protein as detected by NMR, DARTS, and CETSAassays, and our mutagenesis assays also confirmed the essen-tial amino acids for their binding. In addition, we showed thatFTO-high AML samples are more sensitive to CS1 and CS2,while FTO depletion reduced their sensitivity. Collectively,although further systematic studies are warranted to evaluatewhether other reported mechanisms of their actions alsocontribute to the overall anti-cancer efficacy of CS1 and CS2,we have provided compelling evidence that FTO is a direct andessential drug target of both CS1 and CS2. Thus, future clinicaltrials of these two compounds should focus on cancer patientswith a high level of FTO.
Moreover, we showed that FTO is particularly overexpressedin LSCs/LICs, and pharmacological inhibition or KD of FTOsignificantly suppressed LSC/LIC self-renewal. Thus, pharma-cologically targeting FTO holds potent therapeutic potentialbecause it can eradicate LSCs/LICs. FTO’s contribution toLSC/LIC self-renewal is likely through its positive regulation ofMYC and CEBPA (Su et al., 2018), two genes that play importantroles in the maintenance of LSCs/LICs (Li et al., 2014; Ohlssonet al., 2014; Ye et al., 2015; Zhang et al., 2015). FTO KD or inhi-bition also increased the m6A abundance in snRNAs, hinting at arole of FTO in RNA alternative splicing (Mauer et al., 2019; Weiet al., 2018). Similar to FTO, m6A writers (METTL3/14) also targetMYC and play oncogenic roles in AML (Barbieri et al., 2017; Vuet al., 2017; Weng et al., 2018), likely through distinct mecha-nisms (Deng et al., 2018c). Thus, it would be interesting to testwhether targeting both FTO and an m6A writer exhibits a synergyin treating AML.
Evidence is emerging that tumor cells utilize immune check-points as a major mechanism of immune evasion (Beatty andGladney, 2015; Dong et al., 2002). Monoclonal antibodies target-ing the PD-1/PD-L1/PD-L2 axis have achieved encouraging ef-fects in clinical practice in treating multiple types of solid tumors(Gopalakrishnan et al., 2018; Patnaik et al., 2015; Topalian et al.,2012) but have demonstrated only limited effects in AML (Bergeret al., 2008). Here, we show that the expression levels of thesegenes are very low in human AML cells. In contrast, LILRB4,whose activation can promote tumor infiltration and suppressT cell activity (Deng et al., 2018a), is highly expressed in primaryAML. Since the endogenous LILRB4 levels in human primaryAML samples (as well as in AML cell lines) are 40- to 50-foldhigher than those of PD-L1 and PD-L2, LILRB4 appears to bethe major factor that mediates the immune evasion of AML. Inter-estingly, FTO directly upregulates LILRB4 expression via anm6A-dependent mechanism. CS1/CS2 treatment decreasedthe expression of immune checkpoint genes (especially LILRB4)in AML cells, substantially increasing the sensitivity of AML cellsto the cytotoxicity of activated T cells. Different from previousstudies showing the role of YTHDF1 (an m6A reader) in thecross-presentation of tumor antigens and the cross-priming ofCD8+T cells (Han et al., 2019) as well as the role of FTO in promoting melanoma tumorigenesis and anti-PD-1 resistance(Yang et al., 2019), here we demonstrate that by suppressingthe expression of intrinsic immune checkpoint genes (especiallyLILRB4) in AML cells, targeting the FTO/m6A axis substantiallysuppressed immune evasion and sensitized AML cells to T cellcytotoxicity.
In addition, consistent with previous reports (Orskov et al.,2015; Yang et al., 2014), we confirmed that HMA treatment re-sulted in global upregulation of immune checkpoint genes.
Notably, the ascending tendency of LILRB4 is much more signif-icant than PD-L1 and PD-L2 upon DAC treatment. Since the ob-servations that HMA treatment induces upregulation of PD-1/PD-L1/PD-L2 (Orskov et al., 2015; Yang et al., 2014), multipleclinical trials are ongoing now to test therapeutic potential ofthe combinations of HMA with anti-PD-1, PD-L1, or PD-L2 anti-bodies for AML and MDS treatment (Alfayez and Borthakur,2018; Daver et al., 2019; Stahl and Goldberg, 2019). However,as LILRB4 is likely a more critical immune checkpoint genethan the others in AML/MDS, the combinations of FTO inhibitors(or anti-LILRB4 antibody) plus HMAs might be better therapeuticstrategies for AML/MDS treatment. Indeed, we have shown thatFTO inhibitors exhibit strong synergistic effects with HMAs intreating AML in vivo, highlighting the therapeutic potential ofthis combination in treating myeloid malignancies.
Finally, the potent anti-tumor efficacy and minimal side effectsof CS1 and CS2 suggest that that they are highly feasible for clin-ical application. Further studies are warranted to optimize thecompounds to improve their bioavailability, inhibitory effect,and therapeutic efficacy.
Overall, we have identified two potent FTO inhibitors and havedemonstrated that targeting the FTO/m6A axis could signifi-cantly suppress cancer stem cell self-renewal and immuneevasion, highlighting the broad potential of targeting FTOsignaling by effective inhibitors (alone or in combination withother therapeutic agents) for cancer therapy. Moreover, ourFTO inhibitors can also be used as tool compounds for basicand translational research of FTO and m6A modification.