…ζΈοΜ·―ß¬έΈΡ

ΓΓΓΓ’Σ “ΣΘΚΓΓ”Π”ΟΜΖΨ≥DNA(eDNA)ΦΦ θΗΏ–ßΉΦ»ΖΒΊΆξ≥…ΦΪΒΊΥ°…ζΧ§œΒΆ≥(ΦΪ«χ¥σ―σ,ΫϋΑΕΚΘ”ρΦΑΚΰ≤¥ΓΔœΣΝςΒ»)÷–…ζΈοΒΡΈο÷÷Φχ±πΓΔΕύ―υ–‘Ζ÷ΈωΒ»―–ΨΩΙΛΉς,Ε‘ΩΣΖΔΦΪΒΊΒΊ«χ…ζΈοΉ ‘¥ΓΔ‘Λ≤β»Ϊ«ρΥ°…ζΧ§œΒΆ≥±δΜ·«ς ΤΚΆΤΫΚβΈ»Ε®ΚΆ―–ΨΩΦΪΕΥ…ζΨ≥…ζΧ§―ßΈ ΧβΒ»ΨΏ”–÷Ί“ΣΦέ÷ΒΓΘΫϋΡξά¥, eDNAΦΦ θΖΔ’Ι―ΗΥΌ,”––ßΒΊΗΡΫχΝΥ¥ΪΆ≥ΖΫΖ®‘ΎΥ°…ζΧ§œΒΆ≥―–ΨΩ÷–ΒΡ»±Βψ,ΧαΗΏΝΥΕ‘Υ°…ζ…ζΈοΒΡΒς≤ιΙΛΉςΒΡ–ß¬ ΓΘΈΡ’¬Ήέ ωΝΥeDNAΦΦ θΕ‘ΦΪΒΊΥ°ΜΖΨ≥÷–ΒΡ”ψάύΓΔΒΉΤή…ζΈοΓΔΗΓ”Έ…ζΈοΓΔΗΓ”ΈœΗΨζΓΔ≤ΓΕΨΒ»Υ°…ζ…ζΈοΒΡ…ζΈοΕύ―υ–‘Ζ÷ΈωΓΔΈο÷÷ΦχΕ®ΓΔ÷÷»ΚΫαΙΙΖ÷ΈωΦΑ…ζΧ§―ßΒ»ΖΫΟφ―–ΨΩΒΡ”Π”ΟΫχ’ΙΓΘeDNAΦΦ θΉςΈΣ“Μ÷÷–¬–ΥΒΡ…ζΈοΒς≤ιΖΫΖ®,”–ΡήΝΠΦ”ΥΌΗϋ–¬Ζ÷Ή” ±¥ζΒΡœ÷¥ζ…ζΈοΕύ―υ–‘Βς≤ιΒΡΖΫΖ®,‘Ύ―–ΨΩΦΪΒΊΥ°…ζ…ζΈοΕύ―υ–‘…œΨΏ”–ΦΪ¥σΒΡ«ΑΨΑΓΘ
ΓΓΓΓΙΊΦϋ¥ ΘΚΓΓeDNA; ΦΪΒΊ; Υ°ΜΖΨ≥; Υ°…ζ…ζΈο; …ζΈοΕύ―υ–‘;
ΓΓΓΓAbstractΘΚΓΓEnvironmental DNA(eDNA) technology has been applied to efficiently and accurately carry out research works involving species identification and persity analysis of organisms in polar aquatic ecosystems(such as polar oceans, coastal waters, lakes, and streams). This is of great value for the development of biological resources in polar region, prediction of changes in the balance and stability of global aquatic ecosystems, and research on ecological issues in extreme habitats. In recent years, eDNA technology has developed rapidly, effectively addressing the disadvantages of traditional methods in the study of aquatic ecosystems, and improving the investigation of aquatic animals. This review describes progress in the application of eDNA technology in biopersity analysis, species identification, population structure analysis, and other ecological research on aquatic organisms such as fish, benthic organisms, plankton, and viruses in polar aquatic environments. As a new biological survey method, eDNA technology can drastically accelerate the updating of modern biopersity methodologies in the current molecular era, and has infinite prospects in polar aquatic biopersity research.
ΓΓΓΓKeywordΘΚΓΓeDNA; polar; water environment; aquatic organism; biopersity;
ΓΓΓΓ0 ΓΔ“ΐ―‘
ΓΓΓΓΜΖΨ≥DNA(Φρ≥Τe DNA)÷Η÷±Ϋ”¥”ΜΖΨ≥―υΤΖ(»γΆΝ»άΓΔΥ°ΚΆ≥ΝΜΐΈοΒ»)ΜώΒΟΒΡ“≈¥ΪΈο÷ (DNAΤ§ΕΈ)[1,2]ΓΘe DNAΦΦ θ «Ά®Ιΐ¥”ΗςΜΖΨ≥Ϋι÷ ÷–Χα»Γ≥ωΧΊ“λ–‘ΒΡDNA Ε±πΤ§ΕΈ, Ι”ΟDNA≤β–ρΦΦ θΖ÷ΈωΥυΧα»ΓΜΖΨ≥DNAΒΡ Ε±πΤ§ΕΈ«ιΩω,Ε®–‘Μρ’ΏΕ®ΝΩΒΊΖ÷Έω…ζΈοΧε‘ΎΜΖΨ≥Ϋι÷ ÷–ΒΡΨΏΧεΖ÷≤Φ«ιΩωΚΆ…ζΧ§ΙΠΡήΧΊ’ς[1,2]ΓΘe DNAΕ‘‘≠ΈΜ≤…―υ…ζΨ≥÷–ΒΡ…ζΈοάύ»ΚΈό»≈Ε·–‘,“ρ¥ΥΜΙ”–Ω…ΡήΗΡ…ΤΦΪΒΊΥ°…ζΧ§œΒΆ≥ΒΡΜΖΨ≥ΙήάμΚΆΤάΙάΖΫΖ®[3]ΓΘe DNAΦΦ θ‘ΎΥ°…ζΧ§œΒΆ≥Ω…“‘Ϋχ––ΡΩ±ξΈο÷÷ΒΡΦχ±π(»γΆβά¥»κ«÷Έο÷÷ΓΔ±τΈΘΈο÷÷ΚΆ’δœΓΈο÷÷)ΓΔ…ζΈοΕύ―υ–‘Φύ≤β”κΤάΦέΒ»ΙΛΉςΓΘ

ΓΓΓΓ1ΓΔ e DNAΦΦ θΒΡΖΔ’ΙΦρΫι
ΓΓΓΓe DNAΦΦ θΖΔ’Ιάκ≤ΜΩΣDNA≤β–ρΦΦ θΒΡΖΔ’Ι,Ή‘20 άΦΆ70Ρξ¥ζ÷–ΤΎΒΡΒΎ“Μ¥ζ≤β–ρΦΦ θΒ°…ζ“‘ά¥,DNA≤β–ρΦΦ θ“―Ψ≠»ΓΒΟΝΥ÷Ί¥σΫχ’Ι[4,5](ΆΦ1)ΓΘΒΎ“Μ¥ζ≤β–ρΦΦ θ≥ωœ÷‘Ύ1977Ρξ,Φ¥MaxamΚΆGilbertΖΔΟςΒΡΜ·―ßΫΒΫβΖ®ΦΑSangerΒΡΥΪΡ©ΕΥ÷’÷ΙΖ®[6,7]ΓΘΜυ”ΎΒΎ“Μ¥ζ≤β–ρΦΦ θΒΡΖΔ’Ι,2003ΡξΧα≥ωΒΡΧθ–Έ¬κΦΦ θΆ®Ιΐ”ΟΧΊΕ®ΒΡΕΧDNA–ρΝ–ΫΪΈο÷÷Ϋχ––±»Ε‘, Βœ÷ΝΥΕ‘…ζΈοΒΡΖ÷άύΦχΕ®[8]ΓΘΫ”œ¬ά¥”Π‘ΥΕχ…ζΒΡΒΎΕΰ¥ζ≤β–ρΦΦ θ(next-generation sequencing technology,NGS,Φ¥ΗΏΆ®ΝΩ≤β–ρΦΦ θ),ΫβΨωΝΥΒΎ“Μ¥ζ≤β–ρΆ®ΝΩΒΆΓΔΈόΖ®¥οΒΫ¥σΙφΡΘ”Π”ΟΒΡΈ Χβ,ΈΣ…ζΈοΖ÷Ή”―–ΨΩΉω≥ωΝΥΨό¥σΙ±œΉΓΘe DNAΚξΧθ–Έ¬κΦΦ θ‘ΎΕΰ¥ζ≤β–ρΒΡΜυ¥Γ…œΆ®ΙΐΕ‘e DNAΤ§ΕΈΫχ––≤β–ρ,≤ΔΫΪΥυΒΟ–ρΝ–”κ±ξΉΦΧθ–Έ¬κΕ‘±»,ΦχΕ®ΡΩ±ξ…ζΈοΒΡ÷÷άύ[9]ΓΘ1988Ρξ,HandelsmanΒ»[10]Χα≥ωΒΡΚξΜυ“ρΉι―ßΗ≈ΡνΈΣ―–ΨΩΈΔ…ζΈοΧαΙ©ΝΥ–¬ΒΡΥΦ¬ΖΚΆΖΫΖ®ΓΘΒΪ÷±ΒΫΜυ”ΎΒΎΕΰ¥ζ≤β–ρΦΦ θΒΡΚξΜυ“ρΉι―ßΖ÷ΈωΖΫΖ®Ϋ®ΝΔ≤≈ Ι÷±Ϋ”¥”ΜΖΨ≥―υ±Ψ÷–Χα»ΓΜυ“ρΉιDNAΚσΫχ––≤β–ρΖ÷Έω≥…ΈΣΩ…ΡήΓΘΚξΜυ“ρΉι―ßΦΦ θ «Μυ“ρΩΥ¬ΓΈΡΩβΒΡΫχ“Μ≤Ϋ…νΜ·,±ήΟβΝΥΈΔ…ζΈο≈ύ―χΒΡΖ±ΥωΙΐ≥Χ,¥ΌΫχΝΥΕ‘ΦΪΕΥΜΖΨ≥œ¬…ζΈοΕύ―υ–‘ΦΑΙΠΡήΒΡ»œ Ε,‘Ύe DNAΦΦ θΒΡ”Π”Ο÷–Αγ―ί≤ΜΩ…Μρ»±ΒΡΫ«…Ϊ[11]ΓΘΥφΉ≈»ΥΟ«Ε‘DNAΒΡ―–ΨΩΫχ“Μ≤ΫΖΔ’Ι,ΒΎ»ΐ¥ζ≤β–ρΦΦ θ“―≤Μ‘Ό“άάΒΨέΚœΟΗΝ¥ ΫΖ¥”Π(polymerase chain reaction,PCR)ΦΦ θ[12]ΓΘ
ΓΓΓΓΉ‘≤Ϋ»κΜυ“ρΉι―ß ±¥ζ,e DNAΦΦ θΨ≠Ιΐ10”ύΡξΒΡΖΔ’Ι,¥”1987ΡξΩΣ Φ‘ΎΜΖΨ≥ΈΔ…ζΈο―ßΝλ”ρ”Π”Ο,ΈΔ…ζΈο―ßΦ“OgramΒ»[13]≥…ΙΠΒΊ¥”ΚΰΒΉ≥ΝΜΐΈο÷–Χα»Γ≥ωΜΖΨ≥ΈΔ…ζΈοΒΡDNAΓΘ2000Ρξ,“e DNA”ΒΎ“Μ¥Έ‘ΎΈΡœΉ÷–≥ωœ÷,ΗΟ―–ΨΩ÷–άϊ”ΟœΗΨζ»ΥΙΛ»Ψ…ΪΧε(BAC)‘ΊΧεΙΙΫ®ΝΥΜυ“ρΉιDNAΈΡΩβ,≤Δ±μΟςΝΥBACΈΡΩβ÷–Κ§”–Εύ÷÷ΜΖΨ≥DNA,‘Ύ―–ΨΩΆΝ»άΈΔ…ζΈοΕύ―υ–‘ΖΫΟφ”–Ψό¥σΒΡ«±ΝΠ, Ιe DNAΦΦ θ’φ’ΐΒΟΒΫ»œΩ…[14,15]ΓΘ2008Ρξ,e DNAΦΦ θΒΎ“Μ¥Έ‘Υ”Ο”ΎΥ°…ζΧ§œΒΆ≥÷–,―–ΨΩ»Υ‘±άϊ”Ο¥”Υ°―υ÷–Χα»ΓΒΡDNA»ΞΦλ≤β“Μ÷÷‘≠≤ζ”Ύ±±ΟάΒΡ»κ«÷–‘ΝΫΤήΕ·ΈοΟάΙζ≈ΘΆή «Ζώ»κ«÷Υ°”ρ÷–[1]ΓΘ÷°Κσ,e DNAΦΦ θΩΣ ΦΙψΖΚ”Π”Ο”ΎΥ°…ζœΒΆ≥…ζΈο―–ΨΩ÷–ΓΘ2014Ρξ,―–ΨΩ‘±ΒΎ“Μ¥Έάϊ”Οe DNAΦΦ θΦλ≤β»χΩ®άο―«Κ”÷–4÷÷≥ΘΦϊΒΡ»κ«÷”ψάύ,Βς≤ι±μΟςΝΥe DNAΩ…“‘”ΟΉςΦύ≤βΒ≠Υ°…ζΧ§œΒΆ≥÷–»κ«÷”ψάύΒΡ÷Ί“ΣΖ÷Ή”ΙΛΨΏ[16]ΓΘ
ΓΓΓΓΥφΉ≈e DNAΦΦ θΒΡ»’“φ≥… λ,Τδ”Π”Ο”…Ε‘Έο÷÷Φύ≤βΒΡΕ®–‘―–ΨΩΒΫœ÷‘ΎΕ‘…ζΈοΝΩΤάΙάΒΡœύΕ‘Ε®ΝΩΖ÷Έω,―–ΨΩΕ‘œσ”…ΈΔ…ζΈο―–ΨΩΒΫΝΫΤήΕ·ΈοΓΔΒ≠Υ°”ψάύΓΔΚΘ―σ”ψάύΓΔ≈ά––Ε·ΈοΚΆΗΙΉψΕ·Έο―–ΨΩΒ»[17]ΓΘΒΎΕΰ¥ζ≤β–ρΦΦ θΒΡΖΔ’Ι¥Ό Ιe DNAΦΦ θ―ΗΥΌΆΤΙψ,‘ΎΈο÷÷ΦχΕ®―–ΨΩ…œ”…Β±≥θΒΡΒΞΗωΈο÷÷ΦχΕ®ΙΐΕ…ΒΫ’ϊΗω…ζΈο»ΚΒΡΖ÷ΈωΦχΕ®[18]ΓΘ±»»γ,NGSΦΦ θ“―Ψ≠≥…ΙΠ”Ο”ΎΕ‘ΈόΦΙΉΒΕ·Έο(¥σ―υ±Ψ)ΒΡ’ϊΗω»Κ¬δ≤β–ρΙΛΉς÷–[19,20,21,22]ΓΘ¥ΥΆβ, Ι”Οe DNAΩ…Φύ≤β±τΝΌΟπΨχΒΡΒ≠Υ°άΞ≥φΓΔΦΉΩ«άύΕ·ΈοΓΔ”ψάύΚΆ≤Η»ιΕ·Έο,≤ΔΩ…“‘Ά®ΙΐNGSΦΦ θά¥Ϋβ ΆΕ‘’ϊΗωΚΰ«χΒΡΝΫΤήΕ·ΈοΚΆ”ψάύΒΡΦύ≤β«ιΩω[23]ΓΘΖΔ’ΙΒΫ»γΫώΒΡΒΎ»ΐ¥ζ≤β–ρ“―Ψ≠Ω…“‘≤Μ–η“ΣΨ≠ΙΐPCRά©‘ω,ΨΆ Βœ÷ΝΥΕ‘DNAΖ÷Ή”ΒΡ≤β–ρ[24]ΓΘ
ΓΓΓΓΆΦ1 e DNAΖΔ’Ιάζ≥ΧΒΦάάΆΦ
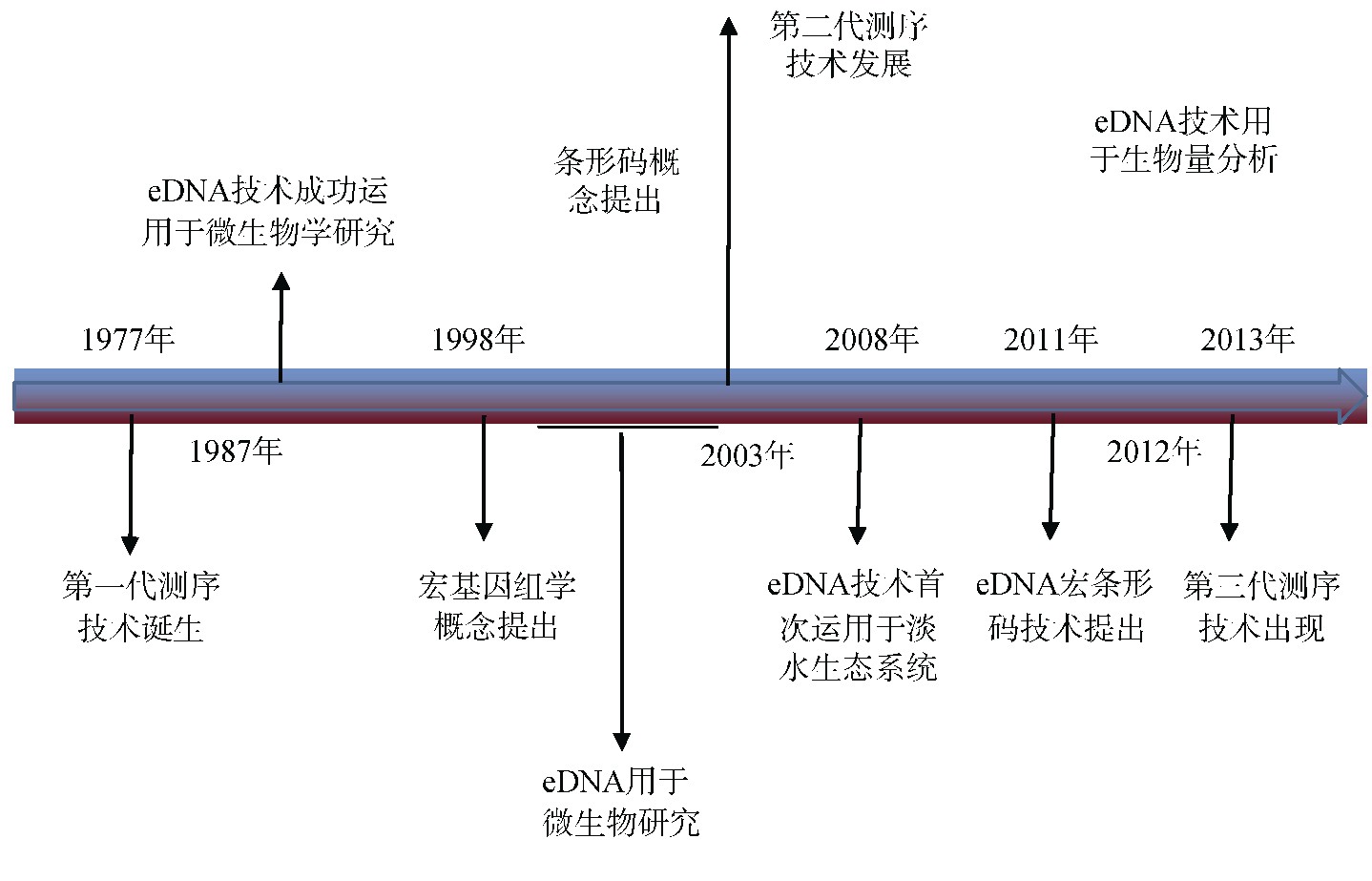
ΓΓΓΓFig.1.Development path of e DNA
ΓΓΓΓΫϋΡξά¥ΫΪ–¬“Μ¥ζe DNAΦΦ θ[24]”κΜζΤς―ßœΑ[25]ΓΔΈά–«“ΘΗ–[26,27]Β»ΦΦ θΝΣ”Ο,“―Ψ≠Ω…“‘¥σ≥ΏΕ»ΓΔΝιΟτΓΔΉ‘Ε·ΒΊΜώ»Γ…ζΧ§Φύ≤β–≈œΔ, Ε±πΥ°…ζΧ§œΒΆ≥÷–Υ°…ζ…ζΈο»Κ¬δΒΡΖ÷≤Φ«ιΩω[28]ΓΘ≥ΐ¥Υ÷°Άβ,e DNAΦΦ θΜΙΨΏ”–“‘œ¬”≈Βψ:”≈‘ΫΒΡΈο÷÷Ω…Φλ≤β–‘ΚΆΧΊ“λ–‘ΓΔ≥…±ΨΫœΒΆ«“≤ΜΜα‘λ≥……ζΧ§œΒΆ≥Η…»≈ΓΔΈό–ηΜώ÷ΣΈο÷÷ΒΡΜυ±Ψ–≈œΔΦ¥Ω…Ϋχ––Φλ≤βΓΔΩ…‘ΎΈόΖ®Ϋχ––¥ΪΆ≥Βς≤ιΒΡΒΊ«χ Β ©Β», ΙΒΟΜΖΨ≥DNAΦΦ θΉςΈΣ“Μ÷÷–¬–ΥΒΡΦύ≤βΖΫΖ®,Υδ»Μ»‘–η“ΣΩΦ¬«ΚΆ±ήΟβ–μΕύœίΎεΚΆ’œΑ≠,ΒΪΗΟΖΫΖ®»‘”–ΡήΝΠΦ”ΥΌΗϋ–¬Ζ÷Ή” ±¥ζΒΡœ÷¥ζ…ζΈοΕύ―υ–‘Βς≤ιΒΡΖΫΖ®,‘Ύ…ζΈοΕύ―υ–‘Φύ≤β―–ΨΩ…œΨΏ”–ΦΪ¥σΒΡ«ΑΨΑ[17]ΓΘ
ΓΓΓΓ2ΓΔ e DNAΦΦ θ‘ΎΦΪΒΊ…ζΧ§œΒΆ≥÷–ΒΡ”Π”Ο
ΓΓΓΓΦΪΒΊΒΊ«χ÷’ΡξΚ°άδΗ…‘ο,±υ―©Η≤Η«,Ηχ‘Λ≤βΚΆΗΡ…ΤΦΪΒΊΜΖΨ≥ΒΡ―–ΨΩΙΛΉς¥χά¥ΦΪ¥σΒΡ≤Μ±ψ,≤Δ«“ΦΪΒΊ…ζΧ§œΒΆ≥ «“ΜΗωΨό¥σΒΡΓΔ«±‘ΎΒΡΒ≠Υ°ΚΆ…ζΈοΉ ‘¥Ωβ, «»Ϊ«ρ…ζΧ§œΒΆ≥ΒΡ÷Ί“ΣΉι≥…≤ΩΖ÷ΓΘΦΪΒΊ…ζΈο «ΦΪΒΊ…ζΧ§œΒΆ≥ΒΡΈο÷ ―≠ΜΖΚΆΡήΝΩΝςΕ·÷–ΒΡ÷Ί“Σ‘ΊΧε,Τδ÷–”–―–ΨΩ±μΟςΦΪΒΊ±υ«χΒΡΚΘ―σ”ψάύΈο÷÷–Έ≥…¬ Οςœ‘ΗΏ”Ύ»»¥χΚΘ―σΒΊ«χ[29]ΓΘΥφΉ≈…ζΈο―–ΨΩΦΦ θΒΡ≤ΜΕœΖΔ’Ι,¥”Ζ÷Ή”ΚΆΜυ“ρΉιΥ°ΤΫΕ‘ΦΪΒΊ…ζΧ§œΒΆ≥ΒΡ―–ΨΩΫΪΖαΗΜΕ‘ΤδΒΡΩΤ―ß»œ÷ΣΓΘ”Π”Ο¥ΪΆ≥–ΈΧ§―ßΖΫΖ®Ε‘ΦΪΒΊΥ°…ζ…ζΈοΫχ––Ε®–‘ΚΆΕ®ΝΩΒΡ―–ΨΩ”–Κή¥σΒΡΨ÷œό–‘,»γ‘≠ΈΜ≤…―υΡ―Ε»Κή¥σ,―υ±ΨΆξ’ϊ–‘Ρ―“‘‘Λ≤βΒ»ΓΘœύΕ‘Εχ―‘,‘ΎΤχΚρΧθΦΰΦΪΈΣΕώΝ”ΒΡΦΪΒΊΒΊ«χ≤…”Οe DNAΦΦ θ≤…―υ ήœό–ΓΓΔΗΏ–ß Γ ±,‘Ύ―–ΨΩΦΪΒΊΥ°…ζ…ζΈοΖΫΟφ”–ΫœΈΣΆΜ≥ωΒΡ”≈ ΤΓΘe DNAΦΦ θœ÷“―ΙψΖΚ”Π”Ο”ΎΦΪΒΊΥ°ΜΖΨ≥÷–”ψάύΓΔΒΉΤή…ζΈοΓΔΗΓ”Έ…ζΈοΓΔΗΓ”ΈœΗΨζΓΔ≤ΓΕΨΒ»≤ΜΆ§άύ»ΚΒΡΕύ―υ–‘Ζ÷Έω”κΤάΙάΓΔΈο÷÷ΦχΕ®ΓΔ»Κ¬δΒΡ…ζΈοΦύ≤βΒ»ΖΫΟφΒΡ―–ΨΩ÷–ΓΘ
ΓΓΓΓ2.1ΓΔ e DNA”Π”Ο”ΎΡœ¥σ―σ”ψάύ―–ΨΩ
ΓΓΓΓΫϋΡξά¥,e DNAΧθ–Έ¬κΦΦ θ±Μ”Π”Ο”ΎΦΪΒΊ”ψάύΒΡ…ζΈοΦύ≤β―–ΨΩ÷–,≤Δ Βœ÷ΝΥΈο÷÷ΦχΕ®ΒΡ±ξΉΦΜ·ΓΘΥυ“άάΒΒΡ≥Θ”ΟΧθ–Έ¬κΜυ“ρΤ§ΕΈΑϋά®œΗΑϊ…ΪΥΊC―θΜ·ΟΗI(COI)Μυ“ρΓΔ“Ε¬ΧΧεΜυ“ρrbc LΓΔ18SΚΥΧ«ΧεRNA(18S ribosomal RNA,18S r RNA)Μυ“ρΓΔΡΎΉΣ¬ΦΦδΗτ«χ(internal transcribed spacers,ITS)Μυ“ρΓΔ16S r RNAΚΆ28S r RNAΒ»ΓΘNearΒ»[30] Ι”ΟœΏΝΘΧε16S r RNAΒΡ»ΪΜυ“ρ–ρΝ–Ε‘Ρœ¥σ―σάύΚζ¬ή≤ΖΥΊ”ψάύPerformformes:NotothenioideiΫχ––œΒΆ≥ΖΔ”ΐ―–ΨΩΖΔœ÷ΡœΦΪ”ψ―«ΡΩ”ψάύΒΡ5Ηω÷ς“ΣΫχΜ·Ζ÷÷ßΖ÷±π «:¬ψΡœΦΪ”ψΩΤΓΔΑΔ œΝζ?ΩΤΓΔΝζ?ΩΤΓΔωυ±υ”ψΩΤΓΔΡœΦΪ”ψΩΤΓΘ
ΓΓΓΓάν‘®Β»[31]άϊ”Ο–ΈΧ§―ßΦχΕ®ΚΆΜυ”ΎœΏΝΘΧεCOIΜυ“ρΒΡe DNAΧθ–Έ¬κΦΦ θΕ‘Ρœ¥σ―σYelcho’Ψ÷ή±ΏΚΘ”ρΒΡ”ψάύΫχ––Έο÷÷ΦχΕ®,¥”≤…Φ·ΒΡ8Έ≤”ψάύ―υΤΖ÷–≥…ΙΠΦχΕ®≥ω3Ηω”––ß÷÷,”–7Έ≤ «ΡœΦΪ”ψΩΤ”ψάύ,1Έ≤¬ψΡœΦΪ”ψΩΤΓΘΫαΚœΝΫ÷÷ΖΫΖ®Ε‘ΡœΦΪ”ψάύΫχ––ΦχΕ®»Ζ±ΘΝΥΦχΕ®ΫαΙϊΒΡΉΦ»Ζ–‘ΚΆ”––ß–‘ΓΘ―–ΨΩ≤ΜΫωΨά’ΐΝΥΧθ–Έ¬κ≤ΈΩΦ ΐΨίΩβ÷–ΒΡ¥μΈσ–ρΝ–,“ΜΕ®≥ΧΕ»…œΜΙΖ¥”≥≥ωΝΥΡœ¥σ―σ”ψάύΒΡΈο÷÷Ήι≥…ΚΆ…ζΈοΝΩΨυ“‘ΡœΦΪ”ψΩΤ”ψάύΈΣ÷ς[31]ΓΘ
ΓΓΓΓ2.2 ΓΔe DNA”Π”Ο”ΎΒΉΤή…ζΈο―–ΨΩ
ΓΓΓΓΒΉΤή…ζΈο «Υ°…ζ…ζΧ§œΒΆ≥ΒΡ÷Ί“ΣΉι≥…≤ΩΖ÷,“ύ «ΚΘ―σΜΖΨ≥÷ ΝΩΒΡ÷Ί“Σ÷Η±ξ,Ε‘ΝΥΫβ…ζΧ§œΒΆ≥ΒΡΫαΙΙΨΏ”–÷Ί“Σ“β“ε[32]ΓΘΑ¥…ζΜνΖΫ ΫΩ…Ζ÷ΈΣΒΉΤή÷≤Έο(ΈΔ‘εΓΔ¥σ–ΆΚΘ‘εΒ»)ΚΆΒΉΤήΕ·ΈοΓΘ“‘ΒΉΤήάΕ‘εΈΣάΐ,άΕ‘ε»Κ¬δ…ζΈοΝΩΜα‘ΎΦΪΒΊΚΰ≤¥ΚΆ≥ΊΧΝΒΡΒΉΤήΜΖΨ≥¥σΝΩ…ζ≥Λ,Ε‘ΙΧΧΦΉω≥ωΝΥ÷Ί“ΣΙ±œΉ[33]ΓΘ
ΓΓΓΓ‘ΎΗΞάΉ»ϊΕϊΚΰΒΉΤήΜΖΨ≥ΒΡ―–ΨΩ÷–ΖΔœ÷”–¥σΝΩΒΡάΕ‘ε…ζΈοΝΩΜΐάέΓΘ Ι”Ο–ΈΧ§―ßΚΆΖ÷Ή”ΖΫΖ®Ε‘ΡœΦΪΈ§Εύάϊ―«÷ίΡœ≤ΩΗΞάΉ»ϊΕϊΚΰΒΡΧλ»ΜΚΆ»ΥΙΛΈΔ…ζΈοΒφ÷–άΕ‘εΒΡ±μ–ΆΚΆΜυ“ρ–ΆΕύ―υ–‘Ϋχ––Ζ÷Έω[34]ΓΘΤδ÷–Ζ÷Ή”ΖΫΖ®÷ΗΑϋά®16S r RNAΜυ“ρΩΥ¬ΓΩβ,±δ–‘ΧίΕ»ΡΐΫΚΒγ”Ψ(denatured gradient gel electrophoresis,DGGE)ΚΆ≤β–ρ[35],Ά§ ±ΫαΚœΙβ―ßœ‘ΈΔΨΒΙέ≤λΫαΙϊ,ΦχΕ®≥ω8÷÷–ΈΧ§–‘;ΕχΖ÷Ή”ΙΛΨΏ‘ρΖΔœ÷ΝΥ15÷÷œΒΆ≥–ΆΓΘΖ÷Ή”ΫαΙϊ±μΟςΡœΦΪάΕ‘εΕύ―υ–‘‘Ε¥σ”ΎΒΞΕά“άΆ–¥ΪΆ≥œ‘ΈΔΨΒΖ÷Έω≥ωΒΡ–ΈΧ§Ζ÷–ΆΓΘ“ρ¥Υ,Ζ÷Ή”ΙΛΨΏΡήΗϋΆξ’ϊΒΊ≤Ι≥δΟη ωΡœΦΪΚΰ≤¥ΒΉΤή÷–ΒΡάΕ‘εΕύ―υ–‘ΓΘ’β“≤ « Ή¥Έ≤…”ΟPCR“ΐΈοΕ‘16S r RNAΜυ“ρΚΆΕ‘άΕ‘ε–ρΝ–ΧΊ“λΒΡITSΫχ––ά©‘ω,≤ΔΫχ––άΕ‘εΕύ―υ–‘Ζ÷ΈωΒΡΕύœύΖ÷ΈωΒΡΆΜΤΤ[34]ΓΘDestombeΒ»[35]≤…”ΟDNAΧθ–Έ¬κΦΦ θΕ‘÷÷‘Ύ≈Ζ÷ό±±¥σΈς―σΚΆΡΠ¬εΗγΚΘΑΕΝΫ÷÷Ϋ≠ίώ τ¥σ–Ά‘εάύΫχ––―–ΨΩ,άϊ”Ο3÷÷ΕάΝΔ±ξΦ«ΒΡΧθ–Έ¬κcox2-cox3ΦδΗτ«χΓΔ“Ε¬ΧΧεΜυ“ρrbc LΚΆITS 2«χ”ρΕ‘Τδ≤ν“λΫχ––“≈¥ΪΖ÷Έω,÷Λ ΒΝΥ±±¥σΈς―σ¥φ‘ΎΝΫΗωΟϊΈΣG.gracilisΒΡ≤Δ––Ζ÷÷ßΈο÷÷“―”–200ΡξΒΡάζ Ζ[35]ΓΘ―–ΨΩΆ§ ±÷ΛΟςΕύΜυ“ρΧθ–Έ¬κΡήΗϋΨΪ»ΖΒΊΟηΜφ’βΝΫ÷÷–ΈΧ§―ßΈο÷÷≤ΔΦλ≤βΦΌΕ®ΒΡ‘”ΫΜΒΡΖΔ…ζΓΘΝθ≥ΩΝΌΚΆΝ÷―ß’ΰ[36]άϊ”Ο–ΈΧ§―ßΚΆDNAΧθ–Έ¬κΕ‘ΑΉΝνΚΘΚΘ”ρΚΆ±υΒΚΗΫΫϋΚΘ”ρΒΡΚ÷‘εΫχ––ΦχΕ®,≤ΔΕ‘Τδά¥‘¥Ϋχ––Ζ÷ΈωΓΘΤδ÷–≤…Ή‘ΑΉΝνΚΘΚΘ”ρΒΡΚ÷‘εΈΣΩΉ“Ε‘ε―«÷÷(Agarum clathratum subsp.Clathratu),ά¥‘¥”Ύ»’±Ψ±±ΚΘΒά,Νμ“Μ÷÷ΈΣΝωΉ¥Ρ““Ε‘ε(Ascophyllum nodosum),≥ΘΦϊ”Ύ±±¥σΈς―σ―ΊΑΕΓΘ
ΓΓΓΓGrantΚΆLinse[37]”Π”ΟDNAΧθ–Έ¬κΦΦ θΕ‘ΡœΦΪΚΘ―σΒΊ«χΑϋά®ΈΛΒ¬ΕϊΚΘΓΔ¬όΥΙΚΘ‘ΎΡΎ °ΦΗΗω«χ”ρΒΡΒΉΤήΈόΦΙΉΒΕ·ΈοΫχ––Έο÷÷ΦχΕ®ΚΆ“≈¥Ϊ―–ΨΩΓΘ―–ΨΩΖΔœ÷’β–©Υ°”ρ÷–ΒΡΈόΦΙΉΒΕ·Έο÷ς“Σ «ΦΉΩ«ΗΌΓΔΜΖΫΎΕ·ΈοΚΆ»μΧεΕ·ΈοΒ»3άύ,ΗΟ―–ΨΩΆ§ ±Ϋ®“ιΡœΦΪΚΘ―σΒΊ«χΜΙ–η“ΣΫχ––Ηϋ…ν»κΒΡDNAΧθ¬κ―–ΨΩ,ά¥ΆΤΕ·ΒΉΤήΕ·ΈοΒΡΖ÷Ή”…ζΧ§―ß―–ΨΩΓΘ
ΓΓΓΓ2.3 ΓΔe DNA”Π”Ο”ΎΗΓ”Έ…ζΈο―–ΨΩ
ΓΓΓΓΗΓ”Έ…ζΈοΩ…Ζ÷ΈΣΗΓ”ΈΕ·ΈοΚΆΗΓ”Έ÷≤ΈοΓΘΆ®Ιΐe DNAΦΦ θ,≤ΜΫω”–÷ζ”ΎΉΦ»ΖΒΊΝΥΫβΗΓ”Έ…ζΈο…ζΧ§ΚΆΙΠΡήΒΡΕύ―υ–‘ΓΔ ±Ω’Ζ÷≤Φ«ιΩω,Ά§ ±ΜΙΩ…Μώ»ΓΈο÷÷Φδ“≈¥ΪΫχΜ·–≈œΔ,―Α’“–¬ΒΡΜυ“ρΙΠΡήΓΘ
ΓΓΓΓΗΓ”ΈΕ·Έο «”…‘≠…ζΕ·ΈοΚΆΚσ…ζΕ·ΈοΉι≥…ΒΡ…ζΈο»Κ¬δ,÷ς“Σ”–‘≠…ζΕ·ΈοΓΔ¬÷≥φΓΔ÷ΠΫ«άύΚΆηψΉψάύΒ»ΥΡάύΓΘΗΓ”ΈΕ·Έο…ζΟϋ÷ήΤΎΕΧ,…ζ≥Λ―ΗΥΌ,Ε‘ΜΖΨ≥±δΜ·ΟτΗ–,≥Θ±Μ”ΟΉς÷Η ΨΈο÷÷ΓΘœύ±»Εχ―‘,ΦΪΒΊΚΘ”ρΒΡΗΓ”ΈΕ·ΈοΒΡ÷÷»Κ―ίΧφ…ν ήΚΘΥ°ΓΔΚΘ±υ±δΜ·ΒΡ”Αœλ,Ε‘”ΎΜΖΨ≥±δΕ·±μœ÷ΒΟΗϋΈΣΟτΗ–«ΩΝ“,≥Θ±Μ”ΟΉς―–ΨΩ»Ϊ«ρΤχΚρ±δΜ·Ε‘ΦΪΒΊ…ζΧ§œΒΆ≥”ΑœλΒΡ÷Ί“Σ÷Η±ξ[38]ΓΘDNAΧθ–Έ¬κΦΦ θ «Ε‘ΦΪΒΊΗΓ”ΈΕ·ΈοΫχ––Έο÷÷ΦχΕ®ΒΡ“Μ÷÷≥Θ”ΟΖΫΖ®ΓΘ
ΓΓΓΓ‘Ύ―–ΨΩΦΪΒΊΗΓ”ΈΕ·Έο…ζΈοΕύ―υ–‘Ζ÷Έω÷–,”Π”Οe DNAΚξΧθ–Έ¬κΦΦ θΩ…“‘ΦΪ¥σΒΊΦρΜ·Ζ÷Έω≤Ϋ÷ηΓΘ≥ΧΖΫΤΫΒ»[40]άϊ”ΟDNAΧθ–Έ¬κΦΦ θ―ι÷ΛΝΥΡœΦΪΚΘ”ρDNAΧθ–Έ¬κ‘ΎΗΓ”ΈΕ·Έο÷÷ΦχΕ®ΒΡ”––ß–‘,―–ΨΩ÷–œΒΆ≥ΒΊ±»ΫœΖ÷ΈωΝΥΡœΦΪΤ’άοΉ»ΆεΚΆΡœΦΪΑκΒΚ÷ή±ΏΚΘ”ρ35÷÷≥ΘΦϊΗΓ”ΈΕ·ΈοΒΡ124ΧθœΏΝΘΧεCOI–ρΝ–[39,40]ΓΘ―–ΨΩΆ§ ±ΖΔœ÷≤ΩΖ÷Έο÷÷ΒΡ÷÷ΡΎ“≈¥Ϊ≤ν“λΥ°ΤΫΫœΗΏ,±μΟςDNAΧθ–Έ¬κΜΙΩ…”ΟΉςœύΙΊΈο÷÷ΒΡ÷÷»Κ“≈¥Ϊ―ß―–ΨΩ[40]ΓΘ¥Υ¥Έ―–ΨΩ–¬‘ωΒΡDNAΧθ–Έ¬κ ΐΨί“‘ΦΑ–¬ΧαΙ©ΒΡΦφ≤Δ“ΐΈοΫΪΆΤΕ·ΡœΦΪΗΓ”ΈΕ·ΈοΜΖΨ≥―υΤΖΒΡΚξΜυ“ρΉι―ß―–ΨΩ[40]ΓΘ‘Ύ±±ΦΪΚΘ”ρΒΡ±»Ϋœ―–ΨΩ÷–“≤÷Λ ΒΝΥDNAΧθ–Έ¬κΦΦ θ‘ΎΦΪΒΊΗΓ”ΈΕ·Έο÷÷άύΦχΕ®ΦΑœύΙΊ―–ΨΩ÷–ΒΡΩ…ΩΩ–‘[41,42]ΓΘChainΒ»[43]‘ΎΕ‘―ΊΦ”ΡΟ¥σΚΘΑΕœΏ(±±ΦΪ)ΙΰΒ¬―ΖΆεΚΆΙΰΒ¬―ΖΚΘœΩΗΓ”ΈΕ·ΈοΒΡ…ζΈοΕύ―υ–‘Ϋχ––¥σΙφΡΘΒΡ ±Ω’ΤάΙά÷–”ΟΒΫΝΥe DNAΚξΧθ–Έ¬κΦΦ θ,―–ΨΩ±μΟς,ΗΟΦΦ θΈΣ¥σΙφΡΘ…ζΈοΕύ―υ–‘Βς≤ιΧαΙ©ΝΥ“Μ÷÷ΦρΜ·ΕχΝιΟτΒΡΖΫΖ®ΓΘHeimeierΒ»[44]Μυ”Ύ16S r RNAΓΔCOIΚΆ18S r RNAΜυ“ρά©‘ωΒΡDNAΧθ–Έ¬κΦΦ θΫαΚœ¥ΪΆ≥–ΈΧ§―ßΖΫΖ®Ε‘ΡœΦΪ¬όΥΙΚΘΦΑΗΫΫϋΥ°”ρΗΓ”ΈΕ·Έο(ΈόΦΙΉΒΕ·Έο”ΉΧε)Ϋχ––ΝΥΈο÷÷ΦχΕ®,÷ς“Σ «Φ§ΤΛΕ·ΈοΓΔ»μΧεΕ·ΈοΓΔ≈Π–ΈΕ·ΈοΚΆΜΖΫΎΕ·ΈοΒ»4ΗωΟ≈ τΓΘ―–ΨΩΆ§ ±ΥΒΟςΝΥ”Π”ΟΖ÷Ή”ΙΛΨΏΩ…ΗϋΚΟΒΊ≤Ι≥δ–ΈΧ§―ßΦχΕ®ΫαΙϊΓΘ
ΓΓΓΓ”κΗΓ”ΈΕ·ΈοœύΥΤ,ΗΓ”Έ÷≤Έο“≤Ε‘ΜΖΨ≥±δΜ·ΦΪΈΣΟτΗ–ΓΘΗΓ”Έ÷≤ΈοΨΏ”–ΗωΧε–ΓΓΔ…ζ≥Λ÷ήΤΎΕΧΓΔ”ΣΗΓ”Έ…ζΜνΒ»ΧΊΒψΓΘΦΪΒΊΈΔ–ΆΗΓ”Έ÷≤ΈοΫœΕύ «ΒΆΒ»ΒΞœΗΑϊ‘εάύ, «ΦΪΒΊ…ζΧ§œΒΆ≥÷Ί“ΣΒΡ≥θΦΕ…ζ≤ζ’ΏΚΆ ≥ΈοΆχΒΡΜυ¥ΓΓΘ άΫγ…œ“―Ψ≠ΖΔœ÷ΝΥ≥§Ιΐ110Εύ÷÷ΦΪΒΊΈΔ–ΆΗΓ”Έ÷≤Έο[45],»ΥΟ«“―Ψ≠ΩΣ Φ”Π”Οe DNAΦΦ θΖ÷ΈωΦΪΒΊΈΔ–ΆΗΓ”Έ÷≤ΈοΕύ―υ–‘[46]ΓΘΗΓ”Έ÷≤ΈοΒΡ÷÷»ΚΫαΙΙΚΆ…ζΈοΝΩΦΑΤδΕύ―υ–‘ΡήΦΑ ±ΒΊΖ¥”≥ΦΪΒΊΥ°”ρ…ζΧ§ΜΖΨ≥ΒΡ±δΜ·[47]ΓΘ¥ΪΆ≥―–ΨΩΗΓ”Έ÷≤ΈοΒΡΕύ―υ–‘ΦΑΖ÷≤Φ«ιΩωΒΡΖΫΖ® «Μυ”Ύœ‘ΈΔΨΒΦΦ θΒΡ–ΈΧ§―ßΖΫΖ®,ΡΩ«Α¥”Μυ“ρΉιΥ°ΤΫ(e DNAΖ÷Έω)Ε‘ΗΓ”Έ÷≤ΈοΫχ––―–ΨΩœύΫœ”Ύ¥ΪΆ≥ΖΫΖ®”–Οςœ‘ΒΡ”≈ Τ[48]ΓΘEilerΒ»[49]άϊ”Ο16S r RNAΜυ“ρΉςΈΣ±ξΦ«ΒΡNGSΦΦ θΚΆœ‘ΈΔΨΒΦΦ θΕ‘ΡœΦΪΦΑ÷ή±Ώ«χ”ρΒΡ49ΗωΚΰ≤¥ΒΡΗΓ”Έ÷≤ΈοΕύ―υ–‘Ζ÷ΈωΖΔœ÷:‘”÷÷Ε·ΈοΟ≈(phylum)Κ§ΝΩΉνΗΏ,Τδ¥Έ «άΕ‘ε(Cyanobacteria)ΓΔ“ΰ‘ε(Cryptophyta)ΓΔ¬Χ‘ε(Chlorophyta)Β»ΓΘ≤Δ«“NGSΦΦ θΒΡΖ÷±φ¬ ΗϋΗΏΓΘ
ΓΓΓΓΕ‘ΜΖΨ≥ΒΡ±δΜ·œλ”ΠΉνΈΣ÷±Ϋ”ΟτΗ–ΒΡ“Μάύ «ΙψΖΚΖ÷≤Φ‘ΎΚΘ±υΓΔΚΰΥ°ΓΔ±υ¥®»ΎΥ°ΚΆΜΐ―©±μ≤ψΒΡΦΪΒΊΈΔ‘εΓΘΡΩ«Α Ι”ΟΜυ“ρΉι―ß≤β–ρΕ‘ΦΪΒΊΈΔ‘εΒΡ―–ΨΩ»‘¥Π”ΎΜυ¥ΓΫΉΕΈ[50]ΓΘΒΎ“ΜΗωΆξ≥…»ΪΜυ“ρΉι≤β–ρΒΡΦΪΒΊΈΔ‘ε «ΡœΦΪΡβ¥ύΗΥ‘ε[51],―–ΨΩΖΔœ÷≤ν“λΒ»ΈΜΜυ“ρ‘Ύ≤ΜΆ§ΜΖΨ≥œ¬±μ¥οΒΡΖαΕ» «≤ΜΆ§ΒΡ,Ά®Ιΐ’β–©Ζ÷ΈωΩ…“‘Ιά≤β–μΕύΦΪΒΊΈΔ‘εΒΡά¥‘¥”κΈ¬¥χ‘εάύ≤ΜΆ§[50]ΓΘ
ΓΓΓΓΗΓ”Έ…ζΈοΒΡΗωΧε¥σ–Γ≤ν“λ¥σ,“άΨί≤ΜΆ§ΝΘΨΕΖ÷ΦΕ,ΗΓ”Έ…ζΈοάύ»ΚΩ…Ζ÷ΈΣΈΔ–Ά“‘ΦΑΈΔΈΔ–ΆΗΓ”Έ…ζΈο,ΥϋΟ«ΧεΜΐΈΔ–Γ,÷÷άύΖ±Εύ,Ρ―“‘Φλ≤β«“–μΕύ÷÷άύ»±ΖΠΟςœ‘ΒΡ–ΈΧ§―ßΧΊΒψΓΘ’κΕ‘±±±υ―σΓΔΡœΦΪΗΫΫϋΥ°Χε÷–ΒΡΈΔΈΔ–ΆΗΓ”Έ…ζΈο,―–ΨΩ’Ώ Ι”ΟΖ÷Ή”…ζΈο―ßΦΦ θΕ‘ΤδΕύ―υ–‘Ϋχ––―–ΨΩ,Μώ»ΓΝΥ¥σΝΩΒΡΈΔΈΔ–Ά’φΚΥΗΓ”Έ…ζΈοΒΡΜυ“ρ–ρΝ–[52,53]Γȸ⑬»[54]άϊ”Οe DNAΦΦ θΕ‘≥ΰΩΤΤφΚΘΚΘ”ρ5Ηω’ΨΈΜ22ΗωΥ°≤ψΒΡ―υΤΖΫχ––ΈΔΈΔ–Ά’φΚΥΗΓ”Έ…ζΈοΒΡ÷÷»ΚΫαΙΙΚΆΩ’ΦδΖ÷≤ΦΖ÷Έω,―–ΨΩ÷–Ά®ΙΐΕ‘ά©‘ωΒΡ18S r RNAΗΏΆΜ±δV4«χΜυ“ρΤ§ΕΈ–ρΝ–Ϋχ––ΗΏΆ®ΝΩ≤β–ρΖ÷Έω,ΒΟ≥ω:ΗςΗΌΒΡΖ÷≤Φ¥φ‘ΎΟςœ‘≤ν“λ,œύΕ‘ΖαΕ»¥σ”Ύ1%ΒΡΈΔΈΔ–Ά’φΚΥΗΓ”Έ…ζΈο”–6ΗωΗΌ,Ζ÷±πΈΣ≤ΜΒ»±όΟΪάύΒΡΫπ‘εΗΌΓΔΙΒ±ό‘εΗΌΓΔ÷–ΙΡ‘εΗΌΓΔ«ύ¬Χ‘εclade IIΓΔ–ΐΟΪΗΌΚΆ“χΕζΗΌ,Τδ÷–Ή‘―χ–ΆΈΔΈΔ–Ά’φΚΥΗΓ”Έ÷≤Έο‘Ύ±±±υ―σΚΘ”ρ‘Φ’Φ85.2%,ΨΏ”–ΨχΕ‘”≈ ΤΓΘ
ΓΓΓΓ2.4 ΓΔe DNA”Π”Ο”ΎΗΓ”ΈœΗΨζ―–ΨΩ
ΓΓΓΓΖ÷≤Φ‘ΎΦΪΒΊΚΘ―σΗςΗωΥ°≤ψΒΡΗΓ”ΈœΗΨζ «ΦΪΒΊ…ζΧ§œΒΆ≥ΒΡ÷Ί“ΣΉι≥…≤ΩΖ÷÷°“Μ,“ΜΕ®≥ΧΕ»…œΈ§≥÷Ή≈ΦΪΒΊΚΘ―σ…ζΧ§œΒΆ≥ΒΡΈ»Ε®–‘ΚΆ…ζΈοΕύ―υ–‘ΓΘ‘ΎΜΖΨ≥―υΤΖ÷–ΖΔœ÷–¬ΒΡ16S r RNAΜυ“ρ–ρΝ–,ΈΣ―–ΨΩ’ΏΟ«―–ΨΩΈΔ…ζΈοΕύ―υ–‘ΧαΙ©“ΜΗω«ΰΒά[55]ΓΘΥϋ «Ά®Ιΐ»ΖΕ®œΗΨζΒΡœΒΆ≥ΖΔ”ΐΙΊœΒΫχΕχΤάΦέΗΓ”ΈœΗΨζΕύ―υ–‘ΒΡΖΫΖ®[56]ΓΘDobsonΒ»[57]”Ύ1993Ρξάϊ”ΟPCRά©‘ωΒΡ16S r RNAΜυ“ρ÷±Ϋ”≤β–ρ≥…ΙΠ Ε±π≥ωΡœΦΪΗΏ―ΈΚΰΝΫΗω–¬Έο÷÷F.gondwanenseΚΆF.salegens,÷Λ ΒΝΥ16S r RNAΜυ“ρ–ρΝ–Ζ÷Έω‘ΎœΒΆ≥ΖΔ”ΐ―–ΨΩ…œΒΡΨό¥σ«±ΝΠΓΘ
ΓΓΓΓZengΒ»[58]Ε‘16S r RNAΜυ“ρΫχ––454ΫΙΝΉΥα≤β–ρΫ“ ΨΝΥ±±ΦΪKongsfjorden(Spitsbergen)ΚΆChukchi BorderlandΝΫΗωΥ°”ρΒΡΗΓ”ΈœΗΨζΒΡΕύ―υ–‘,―–ΨΩΖΔœ÷Kongsfjorden(Spitsbergen)ΒΡΗΓ”ΈœΗΨζ“‘γ-±δ–ΈΨζΚΆΡβΗΥΨζΈΣ÷ς,ΕχChukchi BorderlandΒΊ±μΚΘΥ°―υΤΖ÷–ΒΡ“‘α-±δ–ΈΨζΚΆΖ≈œΏΨζΈΣ÷ςΓΘΆ§ ±2014ΡξZengΒ»[59]άϊ”ΟΆ§―υ454ΫΙΝΉΥα≤β–ρΕ‘ΡœΦΪΑΔΒ¬άΉΆεΦΑ≥Λ≥«ΆεΗΫΫϋ±μ≤ψΚΘΥ°―υΤΖ÷–ΗΓ”ΈœΗΨζΕύ―υ–‘Ϋχ––Ζ÷Έω,ΖΔœ÷ΑΔΒ¬άΉΆε―υΤΖœΗΨζΩ…Μ°Ζ÷ΈΣ18ΗωΟ≈άύ,≥Λ≥«Άε―υΤΖœΗΨζΖ÷ τ”Ύ11ΗωΟ≈άύ,Τδ÷–ΡβΗΥΨζΟ≈ΓΔ±δ–ΈΨζΟ≈ΈΣ”≈ ΤœΗΨζΓΘ
ΓΓΓΓGhiglioneΚΆMurray[60]“‘±ξ«©–ρΝ–ΫΙΝΉΥα≤β–ρΖΫΖ®(Μυ”ΎΨέΚœΟΗΝ¥ ΫΖ¥”ΠΒΡΟΪœΗΙήΒγ”Ψ-ΒΞΝ¥ΙΙœσΕύΧ§–‘ΖΫΖ®ΓΔPCR-DGGEΚΆDGGEΧθ¥χ≤β–ρΓΔPCR±ξΦ«≤β–ρ),Φ¥Ά®Ιΐά©‘ωΗΓ”ΈœΗΨζΒΡ16S r RNAΜυ“ρΒΡΩ…±δV3«χΓΔΗΏ±δV6«χ–ρΝ–Ζ÷ΈωΚΆDGGEΖ÷Έω―–ΨΩΡœΦΪ―ΊΚΘΗΓ”ΈœΗΨζΕύ―υ–‘ΚΆ÷÷»ΚΫαΙΙ±δΜ·ΓΘΆ§ ±Ε‘Ω≠ΕϊΗ«ά »ΚΒΚ(ΩΩΫϋΡœΦΪ)“‘ΦΑΡœΦΪΑκΒΚΗΓ”ΈœΗΨζΫχ––œΒΆ≥–ΆΦχΕ®,ΫαΚœSimpsonΚΆShannon÷Η ΐ‘Λ≤βΈο÷÷ΖαΗΜΕ»ΓΘ―–ΨΩΖΔœ÷:ΝΫΗωΚΘ”ρΒΡΗΓ”ΈœΗΨζ÷ς“Σάύ»Κ±»άΐ”–Οςœ‘≤ΜΆ§,ΒΪΕύ―υ–‘”κ“‘Άυ―–ΨΩœύΥΤ,«“‘ΎΡœΑκ«ρœΡΦΨΒΡΗΓ”ΈœΗΨζΖαΕ»œ‘Ή≈ΗΏ”ΎΕ§ΦΨΓΘCaoΒ»[61]άϊ”ΟΜυ”Ύ16S r RNAΜυ“ρΒΡV1-V3Ω…±δ«χΒΡΫΙΝΉΥα≤β–ρΖ÷Έω―–ΨΩΡœΦΪΑκΒΚ±±ΕΥ±μ≤ψΚΘ”ρΗΓ”ΈœΗΨζ»Κ¬δΫαΙΙΚΆ»Κ¬δΫαΙΙΒΡ”Αœλ“ρΉ”ΓΘ―–ΨΩ÷–Ά®ΙΐΥυΜώΗΓ”ΈœΗΨζΒΡΗ≤Η«Ε»ΓΔΖαΗΜΕ»ΚΆαΕύ―υ–‘÷Η ΐΒ»ά¥ΤάΙάΥ°”ρΗΓ”ΈœΗΨζΕύ―υ–‘,ΖΔœ÷ΉνΖαΗΜΒΡœΗΨζ»Κ «α-±δ–ΈΨζ,Τδ¥Έ «γ-±δ–ΈΨζΓΘ―–ΨΩ÷–Φλ≤βΒΫΒΡΜΖΨ≥“ρΥΊ”Σ―χ―ΈΓΔ“Ε¬ΧΥΊaΓΔ‘≠ΈΜΈ¬Ε»ΚΆ―ΈΕ»Β»÷Η±ξΕ‘ΗΓ”ΈœΗΨζΕύ―υ–‘ΚΆ»Κ¬δ±δΜ·ΒΡ”ΑœλœύΕ‘Ϋœ–Γ[61]ΓΘM?llerΒ»[62]Ά®ΙΐΕ‘16S r RNAΜυ“ρΫχ––ΫΙΝΉΥα≤β–ρΦΑΕ‘≈ύ―χΨζ÷ξΫχ––16S r RNAΜυ“ρ≤β–ρ,―–ΨΩ±±ΦΪΗΏΈ≥Ε»―©÷–ΚΆΒ≠Υ°Κΰ÷–ΒΡœΗΨζ»Κ¬δΫαΙΙΓΘΆ®ΙΐΝΫΗω«χ”ρΒΡœΗΨζΕύ―υ–‘Ζ÷ΈωΩ…÷Σ,‘ΎΦΪΒΊ±υ―©÷–œΗΨζΖαΕ»ΗΏ”ΎΒ≠Υ°Κΰ,ΒΪ «ΦΪΒΊ―©÷–ΚΆΒ≠Υ°Κΰ÷–ΖΔœ÷ΒΡ τ÷°Φδ”–Κή«ΩΒΡ÷ΊΒΰΓΘ
ΓΓΓΓ2.5 ΓΔe DNA”Π”Ο”Ύ≤ΓΕΨ―–ΨΩ
ΓΓΓΓ≤ΓΕΨΙΙ≥…ΝΥΒΊ«ρ…œΉνΖαΗΜΒΡ…ζΈο ΒΧε≤ΔΧαΙ©ΝΥ¥σΝΩΒΡ“≈¥ΪΕύ―υ–‘ΓΘΥϋ «÷Η”Έάκ‘ΎΥ°Χε÷–ΒΡΗς÷÷≤ΓΕΨ, «Υ°…ζΈΔ…ζΈο»Κ¬δΚΆ…ζΧ§œΒΆ≥ΒΡ“ΜΗωΕ·Χ§Ήι≥…≤ΩΖ÷[63]ΓΘΗΓ”Έ≤ΓΕΨΖ÷ΈΣ‘ε≤ΓΕΨΓΔ …ΨζΧεΓΔ …‘εΧε,≥ΐ¥Υ“‘ΆβΜΙΩ…ΖΔœ÷“Μ–©Ε·Έο≤ΓΕΨΚΆ»Υάύ≤ΓΕΨΒ»ΓΘ’κΕ‘≤ΓΕΨΒΡΖ÷Ή”…ζΈοΦΦ θ”–PCR-DGGEΓΔ¬ω≥ε≥ΓΒγ”ΨΦΦ θΓΔΥφΜζά©‘ωΒΡΕύΧ§–‘DNAΨέΚœΟΗΝ¥ ΫΖ¥”ΠΦΦ θΓΔΚξΜυ“ρΉιΈΡΩβΓΔΩΥ¬ΓΈΡΩβΒ»ΓΘ¥”ΚΥΥαΥ°ΤΫ…œ―–ΨΩ≤ΓΕΨΕ·Χ§Ζ÷≤ΦΚΆ“≈¥ΪΕύ―υ–‘,ΦΪ¥σΒΊ¥ΌΫχΝΥΗΓ”Έ≤ΓΕΨ»Κ¬δΫαΙΙΚΆ…ζΧ§―ß―–ΨΩΓΘe DNAΦΦ θΈΣΦΪΒΊΒΊ«χ≤ΓΕΨ»Κ¬δΫαΙΙΚΆ“≈¥ΪΕύ―υ–‘ΧαΙ©“ΜΗω”–«±ΝΠΒΡ–¬ΆΨΨΕΓΘ
ΓΓΓΓ‘Ύ±±ΦΪΚΆΡœΦΪΚΘ±υ÷–ΕΦΙέ≤λΒΫ¥σΝΩΒΡ≤ΓΕΨ[64]ΓΘ≤ΓΕΨ“≈¥ΪΕύ―υ–‘ΒΡ―–ΨΩ «“‘¥σΝΩΒΡœύΙΊ“≈¥Ϊ–≈œΔΈΣ±≥ΨΑΚΆΜυ¥ΓΒΡ,ΡΩ«Α“―÷ΣΒΡ‘ε≤ΓΕΨΓΔ …‘εΧεΒ»ΒΡ»ΪΜυ“ρΉι–ρΝ–ΜΙΫœ…Ό,ΕχΡœΦΪ≤ΓΕΨ―–ΨΩΒΡ÷ς“Σœό÷Τ «»±ΖΠΜυ“ρΉι–ρΝ– ΐΨίΓΘAlbertoΒ»[65]Ά®Ιΐ¥”ΡœΦΪByersΑκΒΚΒΡLimnopolar Lake¥ΩΜ·ΒΡ2 000ΆρΦνΜυΕ‘ΒΡ≤ΓΕΨDNAΫχ––ΫΙΝΉΥα≤β–ρΒΟΒΫ89 347Ηω–ρΝ–,Ζ÷Έω―–ΨΩ≤ΓΕΨ»Κ¬δΒΡΗΏΕ»Εύ―υ–‘,Οη ωΝΥ‘Φ10 000÷÷≤ΓΕΨΜυ“ρ–ΆΚΆœΡΦΨ‘ΎLimnopolar Lake÷–ΖΔ…ζΒΡ≤ΓΕΨΒΡ…ζΧ§―ίΧφ[66]ΓΘ―–ΨΩ±μΟςLimnopolar Lake÷–≤ΓΕΨ»Κ¬δ“‘Η–»Ψ’φΚΥ…ζΈοΒΡ≤ΓΕΨ÷ςΒΦΓΘΆ§ ±Limnopolar LakeΗΏΕ»ΒΡ“≈¥ΪΖαΗΜΕ»±μΟς,‘ΎΤδΥϊάύ»ΚΒΡ…ζΈοΕύ―υ–‘ΫœΒΆΒΡœΒΆ≥÷–,Ω…“‘ΖΔœ÷ΫœΗΏΒΡ≤ΓΕΨΕύ―υ–‘[65]ΓΘΫχ“Μ≤ΫΕ‘Limnopolar LakeΒΡ…ν»κ―–ΨΩ,‘Ύ’ϊΗωΙβΜν‘Ψ‘¬ΖίΚΆ’ϊΗωΡξΕ»÷ήΤΎ÷–ΗϋΤΒΖ±ΒΊΤάΙά≤ΓΕΨ÷÷»Κ,≤ΔΫαΚœΕ‘’ϊΗωΈΔ…ζΈο»Κ¬δΉι≥…ΒΡΦΨΫΎ–‘Ε·Χ§Ϋχ––Ζ÷Έω,ΫΪ≤ΓΕΨ”κΥό÷ςΝΣœΒΤπά¥,ΥΒΟς≤ΓΕΨ‘ΎΡœΦΪΥ°…ζΧ§œΒΆ≥÷–ΖΔΜ”Ή≈÷Ί“ΣΉς”Ο[66]ΓΘ
ΓΓΓΓΚξΜυ“ρΉιΦΦ θ‘Ύ―–ΨΩΦΪΒΊ≤ΓΕΨ…œΩΥΖΰΝΥΕύ ΐΗΓ”Έ≤ΓΕΨ÷÷άύΈόΖ®≈ύ―χΒΡ»±œί,Ρή÷±Ϋ”Ε‘ΜΖΨ≥Μυ“ρΉι–ρΝ–Ϋχ––Ζ÷Έω,¥”ΕχΜώ÷Σ…ζΈοΜυ“ρ–ρΝ––≈œΔ“‘±ψΫχ––Κσ–χ―–ΨΩ[67]ΓΘDanielΒ»[68]Ά®ΙΐΚξΜυ“ρΉι―ßΦΦ θΫβΈω±±ΦΪ6Ηω¥σ–ΆΥ°ΧεΒΡ≤ΓΕΨDNA,Ζ÷ΈωΝΥ±±ΦΪΒ≠Υ°≤ΓΕΨDNA»Κ¬δΫαΙΙΓΘ‘Ύ±Ψ¥Έ―–ΨΩ÷–ΧαΙ©ΝΥά¥Ή‘±±ΦΪΚΆΡœΦΪΒΡDNA≤ΓΕΨΒΡ…νΕ»–ρΝ– ΐΨί,Αϋά®ά¥Ή‘6Ηω±±ΦΪΚΰ≤¥ΒΡ≤ΓΕΨ»Κ¬δ…νΕ»≤β–ρ ΐΨί,≤ΔΫαΚœΕ‘ά¥Ή‘ άΫγΗςΒΊ“ΜœΒΝ–≤ΜΆ§ΒΊάμΈΜ÷Ο…œ“―ΖΔ±μΒΡΒ≠Υ°≤ΓΕΨDNAΫχ––ΝΥ±»ΫœΖ÷ΈωΓΘΫαΙϊ±μΟςΝΥ±±ΦΪΒ≠Υ°≤ΓΕΨ»Κ¬δ”…Έ¥÷ΣΚΆΒΞΝ¥DNA≤ΓΕΨ÷ςΒΦ,Ϋ“ ΨΝΥ“Μ–©ΨΏ”–ΥΪΦΪΖ÷≤ΦΒΡ≤ΓΕΨΤΉœΒΦΑ»Ϊ«ρ…ζΈοΒΊάμΚΆ≤ΓΕΨ»Κ¬δΒΡΝ§Ά®–‘[65];±±ΦΪΒΊ«χ≤ΓΕΨΈο÷÷ΖαΗΜΕ»ΟΜ”–ΫΒΒΆ,ΥΒΟς≤ΓΕΨΩ…Ρή≤ΜΉώ―≠Έ≥Ε»Εύ―υ–‘ΧίΕ»,Ϋ“ ΨΝΥΡœΓΔ±±ΝΫΦΪΈΔ…ζΈο…ζΧ§œΒΆ≥÷°ΦδΒΡ≤ν“λ,…ζΈοΒΊάμ―ßΚΆ≤ΓΕΨ»Κ¬δΒΡΝ§Ά®–‘≤ΜΫωΆΜ≥ωΝΥΦΪΒΊΜΖΨ≥ΒΡΕάΧΊ–‘,“≤ΆΜ≥ωΝΥΝΫΦΪΈΔ…ζΈο…ζΧ§œΒΆ≥÷°ΦδΒΡ≤ν“λ[68]ΓΘBreitbartΒ»[69]ΗυΨίΚξΜυ“ρΉι―ßΕ‘¥”¬μΈ≤‘εΚΘΓΔΡΪΈςΗγΆεΓΔΦ”ΡΟ¥σ≤ΜΝ–ΒΏΗγ¬Ή±»―«ΓΔ±±±υ―σΒΡΫϋΑΕΥ°”ρ ’Φ·ΒΫΒΡΕΧ–ρΝ– ΐΨίΩβΫχ––Ζ÷Έω,―–ΨΩΖΔœ÷ΗΓ”Έ≤ΓΕΨ»Κ¬δάοΚ§”–500ΓΪ130 000ΗωΜυ“ρ–Ά,Τδ÷–Κή¥σ“Μ≤ΩΖ÷Μυ“ρ–Ά‘Ύ≤ΜΆ§ΒΡΒΊάμ«χ”ρ÷–Ι≤Ά§¥φ‘Ύ[70]ΓΘ
ΓΓΓΓ3 ΓΔΫα¬έ
ΓΓΓΓΉέ…œΥυ ω,e DNAΦΦ θ“―ΙψΖΚ”Π”Ο”ΎΦΪΒΊΥ°…ζΧ§œΒΆ≥÷–”ψάύΓΔΒΉΤή…ζΈοΓΔΗΓ”Έ…ζΈοΓΔΗΓ”ΈœΗΨζΓΔ≤ΓΕΨΒΡ―–ΨΩ,≤Μœό”ΎΦΪΒΊ…ζΈο»Κ¬δΕύ―υ–‘ΓΔΈο÷÷ΦχΕ®ΓΔ…ζΈοΦύ≤βΒ»ΩΈΧβΓΘ”»ΤδΜυ”ΎΒΎΕΰ¥ζ≤β–ρΦΦ θΒΡΖΔ’Ι, Ι”Οe DNAΦΦ θΜώ»Γ ΐΨίΕ‘ΦΪΒΊ…ζΈοΫχ––Φύ≤βΖ÷ΈωΒΡΡήΝΠ¥σΖυΧα…ΐ,ΥυΜώ»ΓΒΡe DNA ΐΨίΕ‘ΤάΙά“Μ–©Έο÷÷ΒΡΕύ―υ–‘”–÷Ί“Σ≤ΈΩΦ“β“ε,≤Δ«“ Ι”Οe DNAΦΦ θΡήΖΔœ÷ΚήΕύ–ΈΧ§―ßΡ―“‘ Ε±πΒΡΈο÷÷,‘ΎΖ÷Έω÷÷»ΚΫαΙΙΦΑΤδΉι≥……œΩ…“‘Ζ¥”≥≥ωΗϋΈΣΨΪ»ΖΒΡΫαΙϊ,±»»γΈο÷÷ΒΡ“≈¥ΪΕύ―υ–‘ΓΘΥφΉ≈e DNAΦΦ θΒΡΗΡΫχ,Μυ±ΨΫβΨωΝΥΦΪΒΊΜΖΨ≥…ζΧ§―ß―–ΨΩ…œΖΫΖ®”–œόΓΔ≤…―υΩ’ΦδΨ÷œόΓΔ ΐΨί–≈œΔ¥Πάμ¬ΐΦΑΦύ≤βΫαΙϊ≤ΜΉΦΒ»“ΜœΒΝ–Έ ΧβΓΘΉήΕχ―‘÷°,e DNAΦΦ θ‘Ύ»œ÷ΣΦΪΒΊΒΡΥ°…ζΧ§œΒΆ≥÷–“―Αγ―ί÷Ί“ΣΫ«…Ϊ,”Π”Ο…œ“―ΫΞ«ς≥… λΓΘ
ΓΓΓΓΈ¥ά¥ΖΔ’Ι÷–,Ω…ΫΪe DNAΦΦ θ”κΤδΥϊΦΦ θœύΫαΚœ,“‘”––ß±ήΟβΒΞ“ΜΦΦ θ ΐΨί≤ΜΉΦ»Ζ‘λ≥…ΒΡΤΪ≤νΓΘ±»»γΚξΜυ“ρΉιΈΡΩβ”κΩΥ¬ΓΈΡΩβΫαΚœΖ÷ΈωΡήΗϋΉΦ»ΖΒΊΖ¥”≥ΈΔ…ζΈοΒΡΕύ―υ–‘[67];±»»γ≥ι―υΦλ≤β[71]Ω…ΨΓΩ…Ρή±ήΟβe DNAΦΦ θ‘Ύ≤ΌΉςΙΐ≥Χ÷–≥ωœ÷ΒΡΈέ»ΨΚΆœίΎε,ΧαΗΏΫαΙϊΒΡΉΦ»ΖΕ»Β»ΓΘ≤ΜΕœά©¥σe DNAΦΦ θΒΡ”Π”ΟΖΕΈß,Ε‘ΦΪΒΊΨό¥σΒΡ…ζΈοΉ ‘¥Ϋχ––œΒΆ≥―–ΨΩΆ≥ΦΤΓΔΆξ…Τ≥δ Β ΐΨίΩβΓΔΖΔ’Ι…ζΈο–≈œΔ―ßΙΛΨΏΓΔ‘ωΦ” ΐΨί¥ΠάμΒΡΩ…–≈Ε»,Ω…ΦΧ–χΆΊ’Ιe DNAΦΦ θ‘ΎΦΪΒΊ…ζΈοΕύ―υ–‘Ζ÷ΈωΚΆΜΖΨ≥±ΘΜΛ÷–ΒΡ”Π”ΟΖΕΈß, ΙΤδΫχ“Μ≤Ϋ”Π”ΟΒΫ ≥ΈοΆχΓΔΡήΝΩΝςΕ·ΓΔ÷÷»Κ“≈¥Ϊ–≈œΔ ’Φ·―–ΨΩΒ»ΖΫΟφ[72]ΓΘ
ΓΓΓΓ≤ΈΩΦΈΡœΉ
ΓΓΓΓ[1] FICETOLA G F, MIAUD C, POMPANON F, et al. Species detection using environmental DNA from water samples[J]. Biology Letters,2008, 4(4):423-425.
ΓΓΓΓ[2] HAILE J, FROESE D G, MACPHEE R D E, et al. Ancient DNA reveals late survival of mammoth and horse in interior Alaska[J]. Proceedings of the National Academy of Sciences of the United States of America, 2009, 106(52):22352-22357.
ΓΓΓΓ[3] KELLY R P, PORT J A, YAMAHARA K M, et al. Harnessing DNA to improve environmental management[J]. Science, 2014, 344(6191):1455-1456.
ΓΓΓΓ[4] Ε≠Χλ”ν. DNA≤β–ρΦΦ θ[J].ΦΦ θ”Π”Ο”Ύ―–ΨΩ, 2018(11):71-72.
ΓΓΓΓ[5] –λ ηΟΖ.–¬“Μ¥ζDNA≤β–ρΦΦ θΒΡ”Π”Ο”κ―–ΨΩΫχ’Ι[J].–λ÷ίΙΛ≥Χ―ß‘Κ―ß±®(Ή‘»ΜΩΤ―ßΑφ), 2018, 33(4):60-64.
ΓΓΓΓ[6] SANGER F, NICKLEN S, COULSON A R. DNA sequencing with chain-terminating inhibitors[J]. PNAS, 1977, 74(12):5463-5467.
ΓΓΓΓ[7] MAXAM A M, GILBERT W A. A new method for sequencing DNA[J]. Proceedings of the National Academy of Sciences, 1977, 74(2):560-564.
ΓΓΓΓ[8] HEBERT P D N, CYWINSKA A, BALL S L, et al. Biological identifications through DNA barcodes[J]. Proceedings of the Royal Society of London Series B:Biological Sciences, 2003, 270(1512):313-321.
ΓΓΓΓ[9] POMPANON F, COISSAC E, TABERLET P. Metabarcoding a new way to analyze biopersity[J]. Biofutur, 2011(319):30-32.
ΓΓΓΓ[10] HANDELSMAN J, RONDON M R, BRADY S F, et al. Molecular biological access to the chemistry of unknown soil microbes:A new frontier for natural products[J]. Chemistry&Biology, 1998, 5(10):245-249
ΓΓΓΓ[11] COUNCIL N R. The New Science of Metagenomics:Revealing the Secrets of Our Microbial Planet[M]. Washington DC:National Academics Press, 2007:1-170.DOI:org/10.17226/11902.
ΓΓΓΓ[12] HEATHER J M, CHAIN B. The sequence of sequencers:The history of sequencing DNA[J]. Genomics, 2016, 107(1):1-8.
ΓΓΓΓ[13] OGRAM A, SAYLER G S, BARKAY T. The extraction and purification of microbial DNA from sediments[J]. Journal of Microbiological Methods, 1987, 7(2/3):57-66.
ΓΓΓΓ[14] RONDON M R, AUGUST P R, BETTERMANN A D, et al. Cloning the soil metagenome:A strategy for accessing the genetic and functional persity of uncultured microorganisms[J]. Applied and Environmental Microbiology, 2000, 66(6):2541-2547.
ΓΓΓΓ[15] JO H. Metagenomics:application of genomics to uncultured microorganisms[J]. Microbiology and Molecular Biology Reviews, 2004,68(4):669-685.
ΓΓΓΓ[16] KESKIN E. Detection of invasive freshwater fish species using environmental DNA survey[J]. Biochemical Systematics and Ecology,2014, 56:68-74.
ΓΓΓΓ[17] ΒΞ–ψΨξ,άνΟγ,ΆθΈΑΦΧ.ΜΖΨ≥DNA(eDNA)ΦΦ θ‘ΎΥ°…ζ…ζΧ§œΒΆ≥÷–ΒΡ”Π”Ο―–ΨΩΫχ’Ι[J].”φ“ΒΩΤ―ßΫχ’Ι, 2018, 29(3):23-29.
ΓΓΓΓ[18] REES H C, MADDISON B C, MIDDLEDITCH D J, et al. REVIEW:The detection of aquatic animal species using environmental DNA–a review of eDNA as a survey tool in ecology[J]. Journal of Applied Ecology, 2014, 51(5):1450-1459.
ΓΓΓΓ[19] HAJIBABAEI M, SHOKRALLA S, ZHOU X, et al. Environmental barcoding:A next-generation sequencing approach for biomonitoring applications using river benthos[J]. PLoS One, 2011, 6(4):e17497.
ΓΓΓΓ[20] GIBSON J, SHOKRALLA S, PORTER T M, et al. Simultaneous assessment of the macrobiome and microbiome in a bulk sample of tropical arthropods through DNA metasystematics[J]. Proceedings of the National Academy of Sciences, 2014, 111(22):8007-8012.
ΓΓΓΓ[21] GIBSON J F, SHOKRALLA S, CURRY C, et al. Large-scale biomonitoring of remote and threatened ecosystems via high-throughput sequencing[J]. PLoS One, 2015, 10(10):e0138432.
ΓΓΓΓ[22] THOMSEN P F, WILLERSLEV E. Environmental DNA:An emerging tool in conservation for monitoring past and present biopersity[J]. Biological Conservation, 2015, 183(1):4-18.
ΓΓΓΓ[23] THOMSEN P F, KIELGAST J, IVERSEN L L, et al. Monitoring endangered freshwater biopersity using environmental DNA[J].Molecular Ecology, 2012, 21(11):2565-2573.
ΓΓΓΓ[24] άνΟςΥ§,’‘Οτ.ΒΎ»ΐ¥ζ≤β–ρΜυ±Ψ‘≠άμ[J].œ÷¥ζ…ζΈο“Ϋ―ßΫχ’Ι, 2012(10):1980-1982.
ΓΓΓΓ[25] LIMA-MENDEZ G, FAUST K, HENRY N, et al. Determinants of community structure in the global plankton interactome[J]. Science,2015, 348(6237):1262073.
ΓΓΓΓ[26] TROMAS N, FORTIN N, BEDRANI L, et al. Characterizing and predicting cyanobacterial blooms in an 8-year amplicon sequencing time-course[J].The ISME Journal, 2017, 11(8):1746.
ΓΓΓΓ[27] TORTI A, LEVER M A, J?RGENSEN B B. Origin, dynamics, and implications of extracellular DNA pools in marine sediments[J].Marine Genomics, 2015(24):185-196.
ΓΓΓΓ[28] ’≈Ζ…Νζ,―νΫ≠ΜΣ,―ν―≈ιΣ,Β».ΜΖΨ≥DNAΧθΚξΧθ–Έ¬κΦλ≤βΥ°…ζΧ§œΒΆ≥±δΜ·”κΫΓΩΒΉ¥Χ§[J].÷–ΙζΜΖΨ≥Φύ≤β, 2018, 34(6):37-46.
ΓΓΓΓ[29] RABOSKY D L, CHANG J, TITLE P O, et al. An inverse latitudinal gradient in speciation rate for marine fishes[J]. Nature, 2018,559(7714):392-395.
ΓΓΓΓ[30] NEAR T J, PESAVENTO J J, CHENG C H C. Phylogenetic investigations of Antarctic notothenioid fishes(Perciformes:Notothenioidei)using complete gene sequences of the mitochondrial encoded 16S rRNA[J]. Molecular Phylogenetics and Evolution, 2004, 32(3):881-891.
ΓΓΓΓ[31] άν‘®,’≈»Μ,ΥΈΤ’«λ,Β».Μυ”ΎDNAΧθ–Έ¬κΕ‘ΡœΦΪYelcho’Ψ÷ή±ΏΚΘ”ρ”ψάύΒΡ÷÷άύΦχΕ®[J].ΦΪΒΊ―–ΨΩ, 2018, 30(2):192-197.
ΓΓΓΓ[32] ≤ΧάωΤΦ,ΫπΨ¥Ν÷,Έβ”·Ή”. 2016ΡξœΡΦΨ÷έ…ΫΫϋΑΕΚΘ”ρΒΉΤή…ζΈοΒς≤ι”κ―–ΨΩ[J].ΚΘ―σΩΣΖΔΙήάμ, 2018, 35(9):82-87.
ΓΓΓΓ[33] HAWES I, SCHWARZ A. Photosynthesis in an extreme shade environment:Benthic microbial mats from lake Hoare, a permanently ice-covered Antarctic lake[J]. Journal of Phycology, 1999, 35(3):448-459.
ΓΓΓΓ[34] TATON A, GRUBISIC S, BRAMBILLA E, et al. Cyanobacterial persity in natural and artificial microbial mats of Lake Fryxell(McMurdo Dry Valleys, Antarctica):A morphological and molecular approach[J]. Applied and Environmental Microbiology, 2003,69(9):5157-5169.
ΓΓΓΓ[35] DESTOMBE C, VALERO M, GUILLEMIN M L. Delineation of two sibling red algal species, Gracilaria gracilis and Gracilaria dura(Gracilariales, Rhodophyta), using multiple DNA markers:Resurrection of the species G. dura previously described in the Northern Atlantic 200 years ago[J]. Journal of Phycology, 2010, 46(4):720-727.
ΓΓΓΓ[36] Νθ≥ΩΝΌ,Ν÷―ß’ΰ.ΝΫ÷ξ±±ΦΪΚ÷‘εDNAΧθ–Έ¬κ–ρΝ–ΒΡΦ«¬Φ[J].ΦΪΒΊ―–ΨΩ, 2017, 29(2):228-235.
ΓΓΓΓ[37] GRANT R A, LINSE K. Barcoding Antarctic Biopersity:Current status and the CAML initiative, a case study of marine invertebrates[J]. Polar Biology, 2009, 32(11):1629-1637.
ΓΓΓΓ[38] HAYS G, RICHARDSON A, ROBINSON C. Climate change and marine plankton[J]. Trends in Ecology&Evolution, 2005, 20(6):337-344.
ΓΓΓΓ[39] HEBERT P D N, CYWINSKA A, BALL S L, et al. Biological identifications through DNA barcodes[J]. Proceedings of the Royal Society of London Series B:Biological Sciences, 2003, 270(1512):313-321.
ΓΓΓΓ[40] ≥ΧΖΫΤΫ,ΆθΟτœΰ,ΥοΥ….Μυ”ΎmtCOIΤ§ΕΈ–ρΝ–ΒΡΡœΦΪΚΘ”ρΗΓ”ΈΕ·ΈοΒΡDNAΧθ–Έ¬κ―–ΨΩ[J].ΦΪΒΊ―–ΨΩ, 2014, 26(2):212-221.
ΓΓΓΓ[41] BUCKLIN A, HOPCROFT R R, KOSOBOKOVA K N, et al. DNA barcoding of Arctic Ocean holozooplankton for species identification and recognition[J]. Deep-Sea Research Part I:Topical Studies in Oceanography, 2010, 57(1):40-48.
ΓΓΓΓ[42] ZHANG W W, XIE Y W, YANG J H, et al, Applications and prospects of meta barcoding in environmental monitoring of phytoplankton community[J]. Asian Joumal of Ecotoxicology, 2017, 12(1):15-24.
ΓΓΓΓ[43] CHAIN F J J, BROWN E A, MACISAAC H J, et al. Metabarcoding reveals strong spatial structure and temporal turnover of zooplankton communities among marine and freshwater Ports[J]. Diversity and Distributions, 2016, 22(5):493-504.
ΓΓΓΓ[44] HEIMEIER D, LAVERY S, SEWELL M A. Using DNA barcoding and phylogenetics to identify Antarctic invertebrate larvae:Lessons from a large scale study[J]. Marine Genomics, 2010, 3(3):165-177.
ΓΓΓΓ[45] ΝΚœΰήΩ,ΈΚΕΪ,ΝθΝζΨϋ,Β».ΦΪΒΊΈΔ‘εΒΡ―–ΨΩΫχ’Ι[J].ΚΘ―σΩΤ―ß, 2007,31(4):92-94.
ΓΓΓΓ[46] MOCK T, THOMAS D N. Recent advances in sea-ice microbiology[J]. Environmental Microbiology, 2005, 7(5):605-619.
ΓΓΓΓ[47] Άθ±Η–¬,―νΝΪΖΦ,Νθ’ΐΈΡ.…ζΈοΆξ’ϊ–‘÷Η ΐ”κΥ°…ζΧ§œΒΆ≥ΫΓΩΒΤάΦέ[J].…ζΧ§―ß‘”÷Ψ, 2006, 25(6):707-710.
ΓΓΓΓ[48] EGGE E, BITTNER L, ANDERSEN T, et al. 454 pyrosequencing to describe microbial eukaryotic community composition, persity and relative abundance:A test for marine haptophytes[J]. PLoS One, 2013, 8(9):e74371.
ΓΓΓΓ[49] EILER A, DRAKARE S, BERTILSSON S, et al. Unveiling distribution patterns of freshwater phytoplankton by a next generation sequencing based approach[J]. PLoS One, 2013, 8(1):e53516.
ΓΓΓΓ[50] ΚΈΟάΝ’,≥Ό«…‘Τ,Άθ≥ΛΚΘ.ΦΪΒΊΈΔ‘εΕ‘ΦΪΕΥΜΖΨ≥ΒΡ ”ΠΜζ÷Τ―–ΨΩΫχ’Ι―–ΨΩ[J].ΡœΨ©≈©“Β¥σ―ß―ß±®. 2019, 42(2):201-208.
ΓΓΓΓ[51] FELLER G, GERDAY C. Psychrophilic enzymes:Hot topics in cold adaptation[J]. Nature Reviews Microbiology, 2003, 1(3):200-208.
ΓΓΓΓ[52] LOVEJOY C, MASSANA R, PEDR?S-ALI?C. Diversity and distribution of marine microbial eukaryotes in the Arctic Ocean and adjacent seas[J]. Applied and Environmental Microbiology, 2006, 72(5):3085-3095.
ΓΓΓΓ[53] CHISHOLM S W, FRANKEL S L, GOERICKE R, et al. Prochlorococcus marinus nov. gen. sp.:An oxyphototrophic marine prokaryote containing pinyl chlorophyll a and b[J]. Archives of Microbiology, 1992, 157(3):297-300.
ΓΓΓΓ[54] Έβ‘¬,≤ή ειΣ,ΚΈΫΘΖφ,Β».±±ΦΪ≥ΰΩΤΤφΚΘΈΔΈΔ–Ά’φΚΥΗΓ”Έ…ζΈο»Κ¬δΫαΙΙΦΑΤδΜΖΨ≥œύΙΊ–‘Ζ÷Έω[J].ΦΪΒΊ―–ΨΩ, 2019, 31(1):25-31.
ΓΓΓΓ[55] HUGENHOLTZ P, GOEBEL B M, PACE N R. Impact of culture-independent studies on the emerging phylogenetic view of bacterial persity[J]. Journal of Bacteriology, 1998, 180(24):4765-4774.
ΓΓΓΓ[56] ―Π≥§≤®,ΆθΙζΝΦ,Ϋπ…Κ,Β».ΚΘ―σΈΔ…ζΈοΕύ―υ–‘―–ΨΩΫχ’Ι[J].ΚΘ―σΩΤ―ßΫχ’Ι, 2004, 22(3):377-384.
ΓΓΓΓ[57] DOBSON S J, COLWELL R R, MCMEEKIN T A, et al. Direct sequencing of the polymerase chain reaction-amplified 16S rRNA gene of Flavobacterium gondwanense sp. nov. and Flavobacterium salegens sp. nov., two new species from a hypersaline Antarctic lake[J].International Journal of Systematic Bacteriology, 1993, 43(1):77-83.
ΓΓΓΓ[58] ZENG Y X, ZHANG F, HE J F, et al. Bacterioplankton community structure in the Arctic waters as revealed by pyrosequencing of 16S rRNA genes[J]. Antonie van Leeuwenhoek, 2013, 103(6):1309-1319.
ΓΓΓΓ[59] ZENG Y X, YU Y, QIAO Z Y, et al. Diversity of bacterioplankton in coastal seawaters of Fildes Peninsula, King George Island, Antarctica[J]. Archives of Microbiology, 2014, 196(2):137-147.
ΓΓΓΓ[60] GHIGLIONE J F, MURRAY A E. Pronounced summer to winter differences and higher wintertime richness in coastal Antarctic marine bacterioplankton[J]. Environmental Microbiology, 2012, 14(3):617-629.
ΓΓΓΓ[61] CAO S N, HE J F, ZHANG F, et al. Diversity and community structure of bacterioplankton in surface waters off the northern tip of the Antarctic Peninsula[J]. Polar Research, 2019, 38:1-15.DOI:10.33265/polar.v38.3491.
ΓΓΓΓ[62] M?LLER A K, S?BORG D A, ABU AL-SOUD W, et al. Bacterial community structure in High-Arctic snow and freshwater as revealed by pyrosequencing of 16S rRNA genes and cultivation[J]. Polar Research, 2013, 32(1):17390.
ΓΓΓΓ[63] WOMMACK K E, RAVEL J, HILL R T, et al. Population dynamics of chesapeake bay virioplankton:Total community analysis by pulsed-field gel electrophoresis[J]. Applied and Environmental Microbiology, 1999, 65(1):231-240.
ΓΓΓΓ[64] COLLINS R E, DEMING J W. Abundant dissolved genetic material in Arctic sea ice Part II:Viral dynamics during autumn freeze-up[J].Polar Biology, 2011, 34(12):1831-1841.
ΓΓΓΓ[65] ALBERTO L B, JAVIER T, DAVID V, et al. High persity of the viral community from an Antarctic lake[J]. Science, 2009,326(5954):858-861.
ΓΓΓΓ[66] CAVICCHIOLI R, ERDMANN S. The discovery of Antarctic RNA viruses:A new game changer[J]. Molecular Ecology, 2015, 24(19):4809-4811.
ΓΓΓΓ[67] ΆθΖΦ.±±ΜΤΚΘ‘εάύDNA≤ΓΕΨ“≈¥ΪΕύ―υ–‘―–ΨΩ[D].«ύΒΚ:÷–ΙζΚΘ―σ¥σ―ß, 2017:17.
ΓΓΓΓ[68] DANIEL A C, ALBERTO L B, DAVID A P, et al. Biopersity and distribution of polar freshwater DNA viruses[J]. Microbial Ecology,2015,1:e1400127.
ΓΓΓΓ[69] BREITBART M, FELTS B, KELLEY S, et al. Diversity and population structure of a near-shore marine-sediment viral community[J].Proceedings of the Royal Society B:Biological Sciences, 2004, 271(1539):565-574.
ΓΓΓΓ[70] ANGLY F E, FELTS B, BREITBART M, et al. The marine viromes of four oceanic regions[J]. PLoS Biology, 2006, 4(11):e368.
ΓΓΓΓ[71] JI Y Q, ASHTON L, PEDLEY S M, et al. Reliable, verifiable and efficient monitoring of biopersity via metabarcoding[J]. Ecology Letters, 2013, 16(10):1245-1257.
ΓΓΓΓ[72] ≥¬ΝΕ,ΈβΝ’,Νθ―ύ,–λΚΘΗυ.ΜΖΨ≥DNA metabarcodingΦΑΤδ‘Ύ…ζΧ§―ß―–ΨΩ÷–ΒΡ”Π”Ο[J].…ζΧ§―ß±®, 2016, 36(15):4573-4582