生物化学论文


酶的结构和功能是生物化学的核心问题之一。对于酶催化的化学反应,人们的一般认识是酶通过与底物的相互作用改变反应途径和活化能同等程度地催化正向与逆向反应,不能改变化学平衡的方向和程度[1,2].然而,由于酶结构和功能的多样性与复杂性,酶促反应的化学平衡问题也是复杂的[3,4].现代生物化学研究表明,酶可以通过自身与小分子的结合能改变在酶分子内部的化学平衡,可以通过反应耦合改变外部化学平衡,可以通过动力学控制得到热力学上不利的产物。
因此,“酶促反应不能改变化学平衡的方向和程度”的说法失之过简,在生物化学教学中应适当扩充。同时,认识酶促反应中的化学平衡细节,有助于深入理解酶催化的本质,并指导药物化学设计或构建人工催化体系[5,6].本文结合生物化学和物理有机化学理论,通过若干实例对酶促反应对化学平衡的影响进行分析,以期对教学和研究提供新的思考。
1 酶促反应的内部化学平衡
一个典型的酶促反应经历了以下步骤:酶分子E与底物S结合为复合物E?S;复合物E?S在酶催化下经过过渡态[TScat]≠生成复合物E?P;复合物E?S解离为酶分子E和产物P.ΔG是S生成P的化学反应在没有酶催化条件下的化学反应Gibbs自由能变,对应于外部平衡常数K;ΔGint是酶分子内部S生成P的化学反应Gibbs自由能变,对应于内部平衡常数Kint;结合和解离步骤的Gibbs自由能变分别记为ΔGS和ΔGP;无酶参与时的过渡态[TS]≠与酶结合地过渡态[TScat]≠间的Gibbs自由能差记为ΔGTS.无酶参与的反应活化能记为Ea,酶促反应决速步的活化能记为Eacat,结合和解离步骤的活化能分别记为EaS和EaP.上述各能量之间的关系如图1所示。
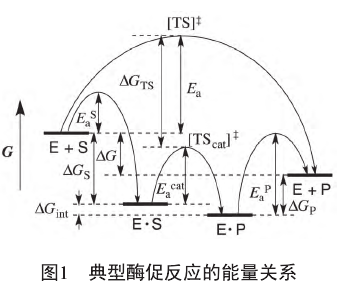
根据诱导契合假说[7],酶促反应的基本原理是通过酶与反应决速步的过渡态[TS]≠的结合将其稳定化为能量更低的[TScat]≠,从而达到降低活化能、提高反应速率的目的。但由于底物、过渡态、产物结构的相似性,酶在与过渡态结构结合的同时势必也要不同程度地结合底物和产物,这使得酶内部底物和产物的能量状态与游离底物和产物的能量状态不同,造成内部化学平衡不同于外部化学平衡。根据图1,ΔGint=ΔG-ΔGS-ΔGP,这说明内部化学平衡取决于酶与底物和产物复合物的相对稳定性。这种相对稳定性会影响酶促反应的速率。根据图1,Eacat=Ea+ΔGTS-ΔGS,如果(ΔGTS-ΔGS)大于0,即酶对底物的稳定化程度超过对过渡态的稳定化程度,就会造成活化能的增大,从而不利于催化反应。如果ΔGP过大,即酶对产物的稳定化作用过强,就会造成EaP过大,同样不利于催化反应。因此,酶对底物和产物的稳定化程度应当维持在合理的水平上。Burbaum等[8]
从理论上证明,为保证酶催化的效率最大,内部化学平衡常数总是趋近于1,ΔGint趋近于0.通过酶与底物、过渡态和产物的结合稳定化作用,酶促反应的内部化学平衡一般与外部化学平衡不同,从而使得酶可以打破热力学限制构建内部反应途径[9],这是酶促反应改变化学平衡的基础。通过多级耦合内部化学反应,酶可以利用外部化学平衡有利的反应贡献的自由能使得外部化学平衡不利的反应得以发生。
2 耦合酶促反应的化学平衡
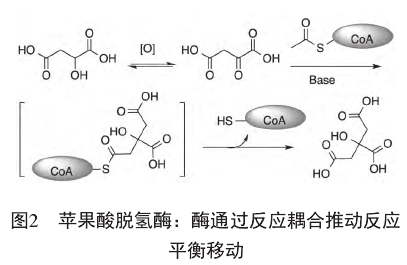
酶可以耦合不同的反应,借此改变平衡方向。要满足这一条件,有两种不同的途径:移走产物或者活化反应物。在移走产物的耦合酶促反应中,酶同时催化两个反应,在第一个反应达到内平衡达到后、E?P释放前就进行第二步反应。总体效果是避免了S与P的直接平衡,而是代之以E?P与E?S的平衡。如果后一步反应的正向进行的趋势很大,就使得第一步反应的平衡发生移动。以苹果酸脱氢酶(malic dehydrogenase)[10]
为例(图2),热力学上苹果酸氧化为草酰乙酸是不利的,但下一步耦合的草酰乙酸与乙酰-CoA的缩合反应、得到的柠檬酸CoA硫酯水解失去CoA-SH的偶联反应都是热力学上十分有利的反应。因此,苹果酸脱氢酶以NAD+为辅因子催化苹果酸氧化为草酰乙酸,并与后续的乙酰CoA缩合、水解硫酯步骤偶联,使得该高度不利的氧化反应得以正向进行。
活化反应物的耦合酶促反应则相当于首先将E?P转化为E?P',进而以新的E?P‘与E?S的平衡取代了S与P的直接平衡。这两步反应同样可以在同一个酶的活性位点中进行,这在自然界也广泛存在[11,12],与化学上的偶联反应很相似。例如羧基直接与氨基作用形成酰胺是热力学熵不利的。但谷氨酰胺合成酶(glutamine synthetase)[13]
可以使用ATP与底物谷氨酸的γ-羧基作用,先形成磷酸活化的羧基,进而被氨分子进攻离去磷酸基团,实现偶联反应形成谷氨酰胺(图3)。这两个反应的净作用是羧酸转化为酰胺,并伴随一分子的ATP水解,利用高能的ATP水解反应来推动羧基的酰胺化反应。可以看出,磷酸活化羧基例子中将羧基与氨基形成酰胺的平衡转化为羧基磷酸与氨基形成酰胺的平衡。
3 酶通过动力学控制改变化学平衡
除了前面提到的热力学手段,酶还可以通过动力学手段影响反应平衡,使得平行反应中的一侧在动力学上加速,另一侧受阻,从而在有限的时间内得到违背平衡产物的动力学控制结果。这种控制是通过酶与平行反应不同过渡态的选择性结合实现的,即特异性地结合过渡态降低某一个方向反应的活化能垒,而难以形成另外一个方向的酶-过渡态复合体,即所谓“定位效应”.以萜类的合成为例,在重要天然产物冰片(Borneol)的生物合成过程中[14,15],环化产物发生碳正离子对双键的亲电加成。在经典有机化学中,卤素对双键的加成遵循马氏规则,即为了得到稳定的碳正离子E2.但是,酶却利用其活性位点的“定位效应”决定反应向反马氏规则E1方向进行。当然,这并不是说酶只能按照反马氏规则进行加成,对于同样的底物,另一种酶则可以实现,并脱去质子得到另一产物蒎烯(Pinene)(图4)。
酶与底物结合后形成的特殊空间构象决定了反应的方向。在重要的单萜Thujone(图5中化合物4)的生物合成中涉及的机理[16,17]的一部分如图5,由三级碳正离子2转化为二级碳正离子3,并进一步水合最终得到酮,这与三级碳正离子更稳定的热力学规则是相违背的。但在这里,酶通过空间效应诱导底物亲核试剂的进攻方向,导致得到的二级碳正离子可以立即转化且对应产物能量更低,相当于通过下一步快速反应的耦合,利用动力学效应左右反应的进行[18,19].
此外,酶从动力学上对反应的调控还受到环境因素的影响。例如,反应介质的改变可以导致酶促反应方向的改变。在含有微量水分的有机溶剂中酶结构发生改变,当其失水造成的动力学刚性和得水造成的热力学不稳定性达到最适条件时,可使酶表现出最高的活性[20].再如,活性位点的“定位效应”受到其中一些关键性作用基团的影响。环境的pH值可能通过影响这些关键性作用基团的电荷状态而极大影响反应平衡。对活性位点的筛选或理性设计可以影响甚至改变反应平衡,乃至改变其底物的识别性能[21].
4 结论
酶促反应中的化学平衡问题是复杂的,酶通过自身与底物、过渡态、产物的特异性结合作用改变化学反应中的能量关系,并达到通过内部化学平衡或动力学控制方法得到非热力学产物的结果,从而在形式上改变了化学平衡。这一点长期在各类教科书上被绝对化地描述为“酶不能改变反应平衡”,这样的认识对于教学与科研是不利的。
参 考 文 献
[1] 王镜岩, 朱圣庚, 徐长法。 生物化学(上册)。 3版。 北京:高等教育出版社, 2002, 320
[2] 傅献彩, 沈文霞, 姚天扬。 物理化学(下册)。 5版。 北京:高等教育出版社, 2006, 292-296
[3] Fersht A. Structure and mechanism in protein science: aguide to enzyme catalysis and protein folding. New York:W. H. Freeman and company, 1999, 103-131
[4] Purich DL. Enzyme kinetics: catalysis & control. Elsevier,2010, 15-25
[5] 马军安, 黄润秋。 酶催化过渡态理论在药物分子设计中的应用。 合成化学, 2000, 8: 479-485
[6] 王志鹏, 冯天师, 崔丽嘉, 等。 过渡态理论于磺胺药物抑菌机理刍议。 中国科学: 生命科学, 2013, 9: 009