生物工程论文

摘 要: 核酸测序技术的诞生是生物医药领域具有里程碑意义的重大突破。以第二代测序为支柱的核酸测序技术在历经十余年的发展与积淀之后,仍在各方面的应用中面临很多挑战。近年来,以第三代测序技术为核心的核酸检测与诊断革新已在生物医药领域崭露头角。第三代测序技术可以实现兆碱基级别超长测序以及无扩增基因组修饰检测,这一系列优势赋予其在传染性疾病的及时诊断、精准医疗、药物研发等领域广泛的应用前景。第三代测序技术可以高效完成病原体的全基因组组装,进而实现病原微生物的全面检测和准确鉴定,此特点对于应对以新型冠状病毒爆发为代表的紧急公共卫生事件至关重要。介绍第三代测序技术的发展、原理及其独特优势,重点阐述第三代测序技术在病原微生物检测以及精准医疗领域的应用以及其在未来生物学领域的价值。
关键词: 第三代测序技术; 病原体检测; 分子诊断; 精准医疗;
2005年,罗氏推出了第一个商业化的二代测序平台——454测序平台[1],结束了以Sanger测序为主导的第一代测序时代[2]。2008年,Solexa/Illumina测序平台和Applied Biosystems SOLi D测序平台紧随其后进入市场[3,4]。与Sanger测序相比,二代测序(second-generation sequencing,SGS)技术可以在保证低成本的情况下,几天之内生成数十万至数十亿个25~800 bp的测序序列数据。凭借高通量的优势,SGS已经占据了大部分的测序市场。但SGS无法实现对DNA准确、全面的直接测序,对天然DNA测序前处理的每一个步骤都可能增加偏好和误差,进而导致测序结果出现错误的风险;样本前期需要经过PCR进行逆转录和扩增,这一过程中GC含量较高的区域无法利用PCR实现高效扩增,加之测序序列读长仍较短,这一系列不足导致其无法实现全基因组覆盖。此外,虽然短读长(short reads)可以较为准确地检测单核苷酸变异(single-nucleotide variations,SNVs),但难以检测和表征结构变异(structural variations,SVs)[5]。

为了解决一代和二代测序技术的局限性,高通量、长读长且能实现无扩增DNA直接测序的第三代测序(thirdgeneration sequencing,TGS)技术逐渐在测序领域崭露头角。TGS能够实现对DNA或RNA的直接测序,减少前期样本的逆转录和扩增步骤,克服读长限制,实现全长测序。同时,TGS可以检测例如DNA的甲基化等基因组修饰。测序技术发展到现在,市场上应用的第三代测序技术主要是太平洋生物科学公司(Pacific Biosciences,Pac Bio)的单分子实时(single-molecule real-time,SMRT)测序技术和牛津纳米孔技术公司(Oxford Nanopore Technologies,ONT)的单分子纳米孔测序技术。本文将以TGS技术的发展和原理为出发点,阐述目前拥有诸多优势的TGS在生物学领域的革新。
1 、第三代测序技术的原理
1.1、 单分子实时测序
如图1所示,SMRT测序技术需要使用双链DNA(double-strand DNA,ds DNA)制备文库[6]:将发夹结构的衔接子连接到DNA分子的两端,形成SMRTbell的环化测序模板[7]。将测序文库加载到SMRT测序单元中,该单元包含纳米级观察室——零模式波导(Zero Mode Waveguides,ZMW)[8]。每个ZMW中的聚合酶引导荧光标记的核苷酸合成待测DNA的互补链,ZMW底部的荧光监测装置实时记录合成时的荧光信号,这些信号生成的测序数据称为连续长读(continuous long reads,CLR)[8]。在测序过程中可以实时检测核苷酸的掺入率,核苷酸掺入之间的时间称为脉冲间持续时间(interpulse duration,IPD),这段时间的长短随DNA表观遗传修饰的出现发生变化[9]。在测序过程中,聚合酶每次持有大约十二个核苷酸,因此单个核苷酸表观遗传水平的变化会影响周围核苷酸的掺入率,这种变化会形成“指纹”[10]。不同的“指纹”对应不同的表观遗传修饰,比如,m6A、m4C和m5C。2011年初,Pac Bio发布了利用SMRT技术发明的Pac Bio RS测序仪,该系统得益于天然高聚合酶的持续合成能力,起初的平均读长相对较短(1.5 kb),平均错误率较高(13%)[11]。随着技术的发展,新测序仪Sequel的平均读长增加了10倍以上,通量增加了100倍;得益于环状模版的构建和聚合酶合成能力的增强,现在可以实现对1~2 kb分子的多次测序,提高了测序准确度。Sequel运行时间短且读长没有受到过多限制,在基因组较短的病原体分子检测方面具备优势[12,13]。该系统还可用于直接鉴定甲基化的碱基,研究核糖体转移RNA动力学,以及进行RNA的直接测序[9,14]。
图1 第三代测序技术原理
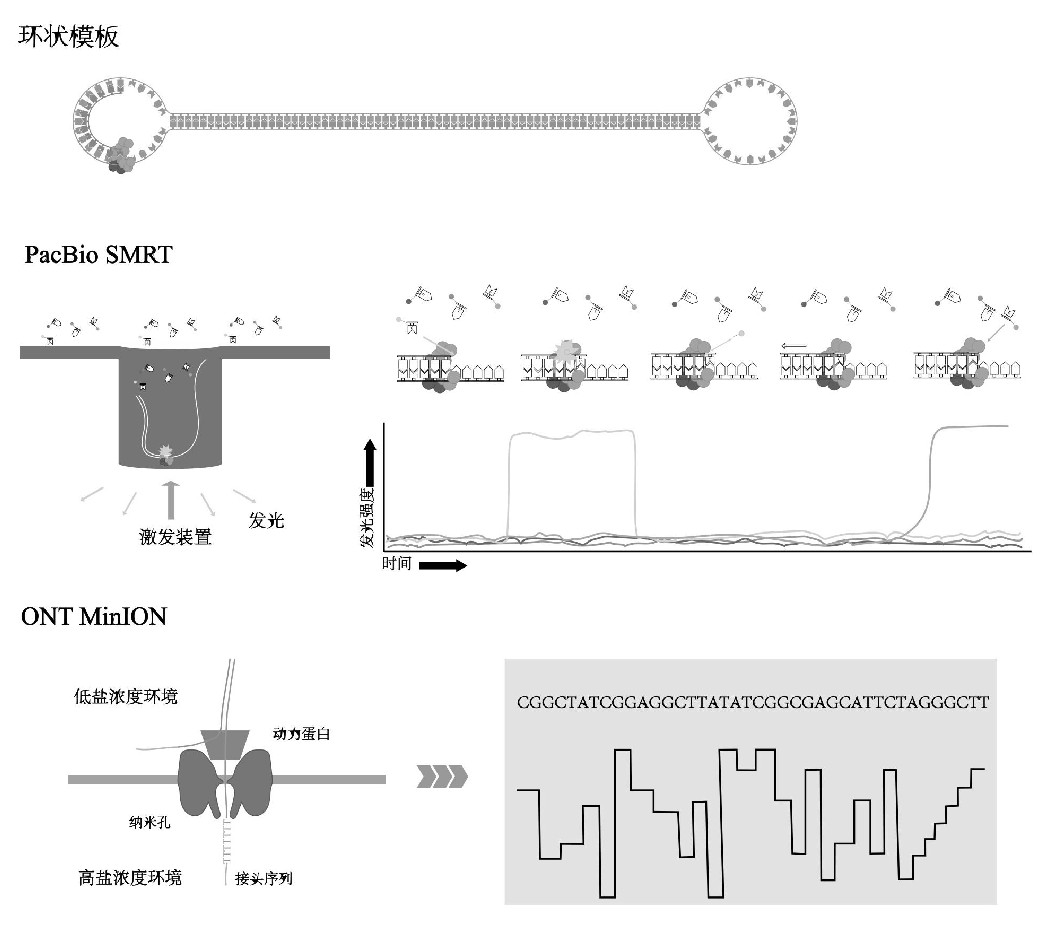
1.2 、纳米孔测序技术
利用纳米孔对单链DNA或RNA分子进行测序的概念起源于20世纪80年代末[14]。将测序衔接子和运动蛋白附着到待测DNA链上,在人工电阻膜上构建生物纳米孔并在膜上施加电压,运动蛋白牵动DNA单链的单个碱基依次通过纳米孔。不同的碱基穿过孔时发生的电流偏转不同,连续相同的碱基会使整个孔的电流变化持续时间变长,以图1所示的检测待测序列的碱基排列。理论上,纳米孔测序技术无需进行任何前期样本处理即可分析极少细胞样本的核酸分子并获得长读数据。除此之外,纳米孔测序还可以直接从RNA甚至是被修饰的核苷酸中获得序列信息。2014年,ONT发布了首款纳米孔测序仪Min ION[15],其具备便携、测序快以及可实现样本全长测序等优点。与SMRT测序相比,纳米孔检测的序列长度不受技术本身的限制,测序的长度主要取决于DNA分子的长度,最大读长可达1 Mb;每条DNA或RNA在纳米孔测序平台中只会被检测一次,该特点可以实现对测序样本的定量;对DNA或RNA的直接测序减少了前期逆转录和扩增步骤,不仅缩短了测序周期,还可实现核苷酸修饰的直接检测。但纳米孔测序无法像SMRT测序一样对同一条链进行多次测序,这造成了其相对较高的测序错误率。据估计,R9.4芯片的纳米孔测序数据错误率在15%左右[16]。为了获得更高的测序准确性,ONT开发了一种针对ds DNA分子进行测序的方法。在ds DNA单端使用发夹衔接子,确保第二条链在第一条链之后进行测序,该系统称为双向(two-directional,2D)测序。该系统目前已经被“1D2”系统取代,“1D2”使用特定序列的衔接子,该衔接子可以提高第一条链测序结束后第二条链继续进入测序孔的概率,这样的改进可以让错误率降低到3%。但因为每个分子的两条链都进行了测序,因此测序通量受到了影响,测序时间也延长了一倍。
2 、第三代测序在生物学领域的革新
2.1 、致病病原体的检测
2.1.1 、病毒的检测
人类一直在寻找可以彻底消灭致病病毒以及及时应对病毒突变的方法。2019年,在湖北武汉发现多起病毒性肺炎病例,该肺炎由严重急性呼吸系统综合症冠状病毒2(severe acute respiratory syndrome coronavirus 2,SARS-Co V-2)引起,世界卫生组织将其命名为COVID-19。首个造成全人类疾病大流行的冠状病毒SARS-Co V-2属于RNA病毒,具备极高的变异速度。早期SARS-Co V-2的研究主要依赖于实时逆转录聚合酶链反应(real-time reverse transcription-polymerase chain reaction,RT-PCR),任何一个病毒检测靶标的突变都会降低现有检测方法的灵敏度。
为确定更稳定的SARS-Co V-2分子检测靶点,实时监测SARS-Co V-2的突变,KELVIN等利用Min ION测序鉴定临床样本中SARS-Co V-2基因组的高表达区域。鉴定后发现位于SARS-Co V-2基因5’端的非结构蛋白1(non-structural protein 1,nsp1)为高表达基因,并以此设计nsp1 RT-PCR验证其在临床检测中的高敏感性和特异性。
在SARS-Co V-2感染人类之前,VIEHWEGER等[17]就尝试利用纳米孔测序对人类肺炎病毒(HCo V-229E)的全长RNA进行直接的检测,观察其结构变体并进行修饰分析。TGS在病毒检测中的应用不只局限于基因组,KIM等[18]利用纳米孔测序技术直接对病毒RNA进行测序,在病毒转录本上找到了至少41个RNA修饰位点,发现经修饰的RNA其poly(A)尾更短,表明RNA修饰与3’ploy(A)尾之间存在联系。在转录组层面,研究发现的未知转录本和RNA修饰的功能可以为理解SARS-Co V-2的生命周期和致病性开辟新的方向。临床应用是实验研究的主要目的,公共卫生紧急事件需要通过实验建立快速的测序报告和详细的流行病学分析。MEREDITH等[19]通过收集5 613例COVID-19患者的临床数据和样本,建立SARS-Co V-2的纳米孔测序诊断技术。通过基因组学分析,在159例患者数据中发现35个相同的病毒簇;经流行病学分析,其中92名患者具有较强流行病学关联。临床、感染控制和医院管理团队通过该诊断技术的反馈结果,可以尽快建立感染控制措施并告知患者安全报告。传统的COVID-19临床诊断方法往往呈现较高的假阴性结果,MING等利用纳米孔测序技术结合靶向扩增开发了纳米孔靶向测序(nanopore targeted sequencing,NTS),在6~10 h内同时检测SARS-Co V-2和其他呼吸道病毒,检测结果显示NTS对SARS-Co V-2的特异性达到100%。在疑似COVID-19病例的61个核酸样本检测结果中,NTS可以灵敏地检测到其中的22个阳性样本;同时NTS可以有效监测突变的核酸序列,对SARA-Co V-2进行分类并检测样本中其他的呼吸道病毒。此外NTS可以进一步扩展,应用到其他病原微生物的诊断当中。综上所述,TGS可以实现对病毒感染样本的全基因组和转录组测序,确定高表达基因区域和结构变体,及时发现高变异病毒的新监测靶点,以便快速阻断高突变病毒的传播。
TGS在病毒检测方面的应用不仅仅局限于检测靶点的发现上,而且可以实现病毒全基因组测序,有助于病毒进化的研究。2014年,Min ION首次应用于西非埃博拉疫情的基因组检测。在资源条件不足的西非,多个团队利用纳米孔测序技术完成了疫情的应急检测和未知病原体的鉴定。2020年,JAN等将PCR、Illumina和Min ION测序相结合,在原有技术上进行改进。无论埃博拉病毒变异与否,该方法均可实现基因组快速精确的检测。结合PCR和TGS,简化后的病毒检测可以从临床样本中成功检测低丰度寨卡病毒基因,进而建立病毒进化树,解析该病毒在感染地的传播途径,为干预病毒传播提供关键数据支撑[20]。同年,STUBBS等[21]利用ONT Min ION测序设备对登革热病毒基因的PCR扩增序列进行测序,可以实现近乎完整的编码区覆盖率。TGS技术在烈性病毒鉴定与进化研究中的应用,极大地解决了以往病毒检测面临的培养周期长、无法定性分析和不能确定未知病原体的问题,对未知病原体及时有效的诊断十分重要。
2.1.2 、寄生虫的检测
致病病原体的概念不仅仅局限在微生物层面,紧急公共卫生事件的始作俑者有时也可能是寄生虫。TGS全长测序的特性可以实现特定致病生物的全基因组测序,进而建立致病病原体耐药性研究体系。寄生虫自身基因组的可变性可以对人类机体的免疫应答迅速做出反应并产生耐药性。比如,疟原虫的耐药性主要是由细胞色素B、Pf CRt、Pf MDR1和K13基因的单核苷酸多态性(single nucleotide polymorphisms,SNPs)和不同突变点之间的组合造成的。为缩短药物研发时间,提高寻找耐药基因的准确度,RUNTUWENE等[22]利用便携的Min ION在野外首次成功鉴定恶性疟原虫的基因分型,利用Min ION鉴定的SNP信息以及K13突变基因,推断出临床疟原虫中的耐药性状态,及时调整药物治疗方案。研究寄生虫的耐药性只是TGS应用的一个方面,TGS也可应用于寄生虫致病机理研究。LIECHTI等[23]利用Min ION对萘氏飞虱的基因组进行测序,通过长读序列的组装和基因修饰得到高质量的基因组,预测可能会在萘氏飞虱发病机理中发挥重要作用的分泌蛋白。经济不发达的国家和地区是寄生虫感染的高发地,由于医疗卫生水平和测序水平较落后,在致病病原体感染暴发初期不能得到有效的控制。以Min ION为代表的TGS测序技术平台具备良好的便携性以及样本处理简易等特点,对于应对需要紧急援助的公共卫生事件至关重要,同时也使野外鉴定病原体基因型成为可能。
2.2 、精准医疗
TGS可以更好地为精准医疗领域提供服务。2001年,人类基因组测序的第一稿完成。在二代测序技术的推动下,经过近二十年改进得到的人类参考基因组(GRCh38)是目前最准确、最完整的脊椎动物基因组[24],但其并未实现端粒到端粒的完整染色体测序。同时,一代和二代测序方法的局限性使人工拼接的染色体存在成百上千的缺口。随着测序技术的发展,高通量、长读长及低成本的TGS可以克服先前测序技术的缺点,使测序数据得以完整地覆盖基因组。利用TGS,加州大学圣克鲁斯分校联合美国国家人类基因研究所(National Human Genome Research Institute,NHGRI)首次合成人类X染色体完整序列,为血友病、慢性肉芽肿病和杜兴式肌肉萎缩症等多种X染色体关联性疾病提供了研究基础[25]。由此可见,基于TGS基因组测序的应用进一步推动了精准医疗的发展。
2.2.1、 生物标志物的探索
精准医疗的应用方向主要集中在癌症治疗研究领域,TGS可应用于预测临床上癌症发生发展的相关生物标志物。急性髓细胞性白血病(acute myeloid leukemia,AML)是一种分子异质性血液系统恶性肿瘤,基因的反复突变是患者治疗和预后反应异质性的原因之一[26,27]。在AML临床研究中,确定具有疾病预测性的生物标志物是开展靶向治疗或进行预后风险临床试验的必要条件。CUMBO等基于纳米孔测序技术建立了更为高效的检测方法,实现了AML中反复突变的六个基因(NPM1、FLT3、CEBPA、TP53、IDH1和IDH2)的快速测序。同样是血液癌症,慢性粒细胞白血病(chronic myrloid leukemia,CML)则是由9号染色体和22号染色体之间的易位产生BCR-ABL1融合蛋白引起的病变。临床中,CML患者通常通过酪氨酸激酶抑制剂(tyrosine kinase inhibitors,TKIs)进行治疗,但该疗法可诱导靶基因点突变导致耐药性。CAVELIER等[28]构建了一个约1.5 kb的BCR-ABL1 c DNA扩增子,并利用SMRT进行测序检测TKI的抗性突变,检测阈值远远低于Sanger测序。以癌症为例,罹患同类癌症的不同病人致病基因具有异质性。TGS能够实现病患生物标志物的个性化研究,推进了个性化精准医疗的应用和发展。
2.2.2、 人类表观遗传机制的探索
癌症的发生通常伴随着基因组表观遗传学修饰的改变。TGS利用电信号进行测序的原理赋予其直接检测碱基修饰的能力,为探索表观遗传学机制奠定了基础。表观遗传学机制虽不涉及基因组核苷酸的改变,但可通过表观遗传修饰介导下游基因表达模式发生改变,此现象可跨代遗传。与单纯研究遗传因素相比,探索表观遗传学机制是解析疾病致病机理的又一重要研究方向。在1型糖尿病(diabetes mellitus type1,T1D)中,表观遗传修饰异常(DNA甲基化、组蛋白修饰和micro RNA失调)与致病基因表达的改变息息相关[29]。目前T1D主要的易感基因是HLA II类等位基因,具有50%的遗传风险。已有研究表明,TGS与靶向捕获技术联用可实现人类白细胞抗原(human leukocyte antigen,HLA)的高分辨基因分型及主要组织相容性复合体(major histocompatibility complex,MHC)区域单倍体类型的精确鉴定[30]。SUZUKI等[31]利用SGS和TGS进行全长HLA等位基因测序,通过对46名健康受试者的基因组进行测序,检测到253个不同的等位基因,其中137个是新颖等位基因。基因组等位基因分辨率的提高增加了基因多态性,为精准医疗和HLA相关疾病的研究提供理论依据。TGS节省了样本前期的PCR过程,将原始基因组的表观遗传修饰完整地保留了下来,并利用电信号测序或荧光脉冲间时间的长短实现基因组修饰的检测,这一点是SGS无法企及的。因此,TGS实现未扩增样本的测序不仅可以减少偏差,同时为表观遗传修饰的研究提供了可能。
3、 展望
TGS克服了Sanger测序和SGS在样本处理、测序通量和测序读长上的限制。读长的增加减少了基因组拼接的成本和时间,避免了PCR扩增时可能引入的错误,同时能够实现表观遗传中DNA的修饰和基因结构变异方面的研究。此外,TGS也可以完成药用植物的全基因组拼接,解析和健全药用植物的分子作用机制。单萜吲哚生物碱(monoterpene indole alkaloid,MIA)包含3 000多种不同化学骨架的化合物,是中草药中重要的有效成分,主要通过化学方法从长春花、夹竹桃和喜树中提取。鉴于MIA复杂的化学性质及其在药用上的价值,需要研究MIA的基因簇在不同植物中是否一致,以及在不同的植物中是否存在加速MIA生物合成的分子。钩吻是产生MIA的代表性植物之一,LIU等[32]借鉴棉花和人类基因组组装的成功经验,使用纳米孔测序技术和高通量染色体构象捕获技术(high-throughput chromosome conformation capture techniques,Hi-C)组装钩吻基因组,研究合成MIA的基因序列,拓展中药在临床治疗中的用途。
TGS的检测速度和检测长度的优势可以在紧急疫情爆发时发挥关键作用,符合国家卫健委在《关于全面推进社区医院建设的通知》中将强化传染病早发现、早报告能力作为社区医院的主要建设任务之一的宏观指导。花最少的时间获得最多的候选病原体信息是快速应对公共突发疫情的前提。中国疾病预防控制中心分别以Ion Torrent PGM、Pac Bio SMRT Sequal和ONT Min ION这三个测序平台为基础建立病毒检测工作流程(virus identification pipeline,VIP)[33]。该方案解决了TGS应用到病毒检测领域时所面临的两个重要问题:(1)是否可以从复杂的临床样本中准确地识别出病毒;(2)能否完整覆盖病毒全基因组。同时,通过比较不同的测序平台数据得出结论:Sequal能够准确覆盖病毒基因组,Min ION可以使用最低的成本实现最快的测序速度,尽管准确度相对较低,但能够完成病毒分类鉴定。这类流程的建立,推动TGS快速准确地应对致病病原体的传播。综上所述,TGS不仅可以在传染性疾病的及时诊断、精准医疗、药物研发等领域广泛应用,而且在中草药有效成分的合成生物学领域具有重要的指导意义。第三代测序技术的应用如图2所示。
目前的TGS技术同样存在不足:(1)在实现DNA或RNA的全长测序时,对样本本身的核酸质量有很高的要求,测序前样本的抽提必须尽可能减少人为造成的误差;(2)TGS的测序错误率仍然相对较高,这也是目前的TGS技术无法真正替代SGS的原因。但值得庆幸的是,测序技术的不断改进和算法的助力正在尝试解决TGS面临的瓶颈,帮助进一步拓展其在生物学领域的应用。
图2 第三代测序技术的应用
参考文献
[1] MARGULIESL M,EGHOLML M,ALTMAN W,et al.Genome sequencing in microfabricated high-density picolitre reactors[J].Nature,2005,437(7057):376-380
[2] SANGER F,COULSON A R.A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase[J]J mol biol,1975,94(3):441-448.
[3] BENTLEY D R,BALASUBRAMANIAN S,SWERDLOW H P,et al.Accurate whole human genome sequencing using reversible terminator chemistry[J].Nature,2008,456(7218):53-59.
[4] VALOUEV A,ICHIKAWA J,TONTHAT T,et al.Ahigh-resolution,nucleosome position map of C.elegans reveals a lack of universal sequence-dictated positioning[J].Genome res,2008,18(7):1051-1063.
[5] SALK J J,SCHMITT M W,LOEB L A.Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations[J].Nat rev genet,2018,19(5):269-285.
[6] EID J,FEHR A,GRAY J,et al.Real-time DNA sequencing from single polymerase molecules[J].Science,2009,323(5910):133-138.
[7] TRAVERS K J,EID J S,RANKET D R,et al.A flexible and efficient template format for circular consensus sequencing and SNP detection[J].Nucleic acids res,2010,38(15):159.
[8] RHOADS A,AU K F.Pac Bio sequencing and its applications[J].Genomics proteomics bioinformatics,2015,13(5):278-289.
[9] FLUSBERG B A,WEBSTER D R,LEE J H,et al.Direct detection of DNA methylation during single-molecule,real-time sequencing[J].Nat methods,2010,7(6):461-465.
[10] SCHADT E E,BANERJEE O,FANG G,et al.Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases[J].Genome res,2013,23(1):129-141.
[11] QUAIL M A.A tale of three next generation sequencing platforms:comparison of Ion Torrent,Pacific Biosciences and Illumina Mi Seq sequencers[J].BMC genomics,2012(13):341.
[12] CHIN C S,SORENSON J,HARRIS J B,et al.The origin of the Haitian cholera outbreak strain[J].N engl j med,2011,364(1):33-42.
[13] RASKO D A,WEBSTER D R,SAHL J W,et al.Origins of the E.coli strain causing an outbreak of hemolyticuremic syndrome in Germany[J].N engl j med,2011,365(8):709-717.
[14] DEAMER D,AKESON M,BRANTON D.Three decades of nanopore sequencing[J].Nat biotechnol,2016,34(5):518-524.
[15] JAIN M,FIDDES I T,MIGA K H,et al.Improved data analysis for the Min ION nanopore sequencer[J].Nat methods,2015,12(4):351-356.
[16] JAIN M,TYSON J R,LOOSE M W,et al.Min IONanalysis and reference consortium:phase 2 data release and analysis of R9.0 chemistry[J].F1000Res,2017(6):760.
[17] VIEHWEGER A,KRAUTWURST S,LAMKIEWICZ K,et al.Direct RNA nanopore sequencing of full-length coronavirus genomes provides novel insights into structural variants and enables modification analysis[J].Genome res,2019,29(9):1545-1554.
[18] KIM D,LEE J,YANG J,et al.The architecture of SARS-Co V-2 transcriptome[J].Cell,2020,181(4):914-921.
[19] MEREDITH L W,HAMILTON W L,WARNE B,et al.Rapid implementation of SARS-Co V-2 sequencing to investigate cases of health-care associated COVID-19:a prospective genomic surveillance study[J].Lancet infect dis,2020,3099(20):30562-30564
[20] GRUBAUGH N D,LADNER J T,DUDAS G,et al.Genomic epidemiology reveals multiple introductions of Zika virus into the United States[J].Nature,2017,546(7658):401-405.
[21] STUBBS S C B,BLACKLAWS B A,YOHAN B,et al.Assessment of a multiplex PCR and nanopore-based method for dengue virus sequencing in indonesia[J].Virol j,2020,17(1):24.
[22] RUNTUWENE L R,TUDA J S B,MONGAN A E,et al.Nanopore sequencing of drug-resistance-associated genes in malaria parasites,plasmodium falciparum[J].Sci rep,2018,8(1):8286.
[23] LIECHTI N,SCHURCH N,BRUGGMANN R,et al.Nanopore sequencing improves the draft genome of the human pathogenic amoeba naegleria fowleri[J].Sci rep,2019,9(1):16040.
[24] LANDER E S,LINTON L M,BIRREN B,et al.Initial sequencing and analysis of the human genome[J].Nature,2001,409(6822):860-921.
[25] MIGA K H,KOREN S,RHIE A,et al.Telomere-totelomere assembly of a complete human X chromosome[J].Nature,2020,585(7823):79-84.
[26] ARBER D A,ORAZI A,HASSERJIAN R,et al.The2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia[J].Blood,2016,127(20):2391-2405.
[27] DOHNER H,ESTEY E,GRIMWADE D,et al.Diagnosis and management of AML in adults:2017 ELNrecommendations from an international expert panel[J].Blood,2017,129(4):424-447.
[28] CAVELIER L,AMEUR A,CAHILL N,et al.Clonal distribution of BCR-ABL1 mutations and splice isoforms by single-molecule long-read RNA sequencing[J].BMCcancer,2015(15):45.
[29] DANG M N,BUZZETTI R,POZZILLI P.Epigenetics in autoimmune diseases with focus on type 1 diabetes[J].Diabetes metab res rev,2013,29(1):8-18.
[30] CHEN J,SHU M Y,LI J,et al.The third-generation sequencing combined with targeted capture technology for high-resolution HLA typing and MHC region haplotype identification[J].Yi chuan,2019,41(4):337-348.
[31] SUZUKI S,RANADE S,OSAKI K,et al.Reference grade characterization of polymorphisms in full-length HLAclassⅠandⅡgenes with short-read sequencing on the IONPGM system and long-reads generated by single molecule,real-time sequencing on the pacbio platform[J].Front immunol,2018(9):2294.
[32] LIU Y,TANG Q,CHENG P,et al.Whole-genome sequencing and analysis of the chinese herbal plant gelsemium elegans[J].Acta pharm sin b,2020,10(2):374-382.
[33] LI Y,HE X Z,LI M H,et al.Comparison of third-generation sequencing approaches to identify viral pathogens under public health emergency conditions[J].Virus genes,2020,56(3):288-297.