ҩѧ����

����ժ Ҫ������ҩƷ�����Ŀ����DZ�֤ҩƷ��ȫ��Ч����Ҫ��ʩ��Ҳ����������ҩƷ�����Ĺؼ����ڡ���2010�����ʵʩ�������ƵĻ�����������������ʮ�������Ŭ��, �����Ѿ��γ���һ���Ƚϳ����ҩƷ����������ϵ����������2010��֮ǰ��2010~2015��仯ѧҩƷ�������ƵĽ�չ������������2015�����������������������������������������Ӧ�õȷ��������Ѹ�ٷ�չ����˱�������2015��������ѧҩƷ���ʿ��ƵĽ�չ�����������ؽ�����������ͷ�չǰ����
�����ؼ��ʣ���������; ��ѧҩƷ; ���ʼ���; ��������; ҩƷ��;
����Abstract����Impurity profiling is one of the most important activities in both assuring drug safety and improving the quality of domestic drugs. Since the basic strategy of impurity profile control was put forward in 2010, a mature control procedure for impurity profile in drugs has been formed in China after nearly ten years of continuous efforts. The progress in impurity profiling before 2010 and from 2010 to 2015 have been reviewed. Since 2015, the concepts, analytical techniques and the application of these techniques in this field have developed rapidly. As a result, the progress in impurity profiling of chemical drugs since 2015 was reviewed in this paper. And the views on future development of impurity profiling in drugs were also put forward.
����Keyword����impurity profile; chemical drug; impurity identification; impurity assessment; pharmaceutical standard;
������ҩ�������ף�impurity profile���Ŀ����DZ�֤ҩƷ��ȫ�Ե���Ҫ���ڣ�Ҳ��Ŀǰ������ҩ�з��Ĺؼ���Լ���ء�������������صĹؼ���������ɸ���Ϊ��������ϵ�����ķ������������ֵĽṹ����������ֵĶ���������������[1]�������“��������(impurity profiling)”����Ӧ���ҩƷ�е�ÿһ�����ʣ����������������ƶ���Ӧ���ʿ��ȡ��ڹ����ش���ҩ���Ƶ���Ŀ��֧���£������������������Ƽ�������Ѹ�ٷ�չ����������2010—2015��仯ѧҩƷ�������о���չ���й�����[2]������������2015��������ѧҩƷ�������о��Ľ�չ��
����1�� ���桢ָ��ԭ����Ӧ��
��������ҩƷע�Ἴ��Ҫ�����Э���ᣨICH�����ƶ���ԭ��ҩ���Ƽ������о�ָ��ԭ��ICH Q3A��ICH Q3B���������ܼ��о�ָ��ԭ��ICH Q3C����Ԫ�������о�ָ��ԭ��ICH Q3D���Ļ����ϣ�2014���ְ䲼�˻����������о�ָ��ԭ��[Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to limit Potential Carcinogenic Risk��ICH M7 (R1)]����һ��ָ�����»�ѧҩ�з��е������о�����ȻICH��ָ��ԭ�����ҩע��ʱҩƷ�еĸ�������������ȷ��Ҫ����ν�ָ��ԭ���������з�ʵ�������������������Ҫ̽�֡�
����ҩƷ�е����ʿ�����Դ��ԭ�Ϻϳ��е���ʼ��ܼ����������м��塢������ȹ��չ��̣�Ҳ�������Ƽ������������ʹ�ù����в�������ҩƷ���з�����Ʒ����ͨ��Ҫ�����ϳ���ʱ�䣬��ͬ�з��εĹ�ע�ص�Ӧ������ͬ�������ǶԲ�Ʒ�����ʵ���ʶҲ�ǰ����ŶԲ�Ʒ���ա�����������IJ�����֪�������˽⡣Ȼ����Ŀǰ������ʳƷҩƷ�ල�����֣�FDA����ŷ��ҩƷ�����֣�EMA�������ҩ��ͬ�з��������о��Ĺ�ע����й�ԭ���Ե����ۡ�Olsen��[3]�Դ˽��������������ڹ������ʣ�Ӧ��ԭ�Ϻϳɽ��ص��ע��Ʒ�п��ܳ��ֵĸ���DZ�����ʣ������պϳ�·��ȷ����Ӧ�ص�������ʵ�ȥ��;����ȷ�����������еĹؼ��ʿص㣻���Ź��չ��̵IJ��ϳ��죬�ٿ�չδ֪���ʵĽṹȷ�Ϲ������������µķ�������ȷ���Ƿ���DZ�����ʵĴ��ڡ������������ʣ�ͨ����Ӻϳɹ��յĽǶȿ��Ƹ��������칹��IJ���������ҩ�з������ڣ����ʵ�ˮƽ���Ʒ�Ķ���ѧ��ȫ�����۽����ƥ�䣻����ҩ�����ٴ��о��Σ���Ʒ�е������ȿ���ICH��Ҫ����п��ƣ�Ҳ���Ի����ٴ���¶�����Ͷ���ѧ����ʶȵ�������ʱ�����ڿصİ�ȫ�����ߣ����ʵļ�����ֵ�ͽ綨��ֵ���Ե�����ICH Q3��3�����������ٴ���¶�����ı仯������Ӧ�ĵ���[4]��������III���ٴ�ʱ����ƷӦ����ICH��Ҫ��
����ICH M7 (R1) ��ҩƷ�еĻ��������ʣ�mutagenic impurities, MIs���Ѿ�����ȷ�Ŀ���Ҫ�����ݶ���ѧ��ע��ֵ(TTC)��������������Ϊ1.5 μg����������ձ���Ϊ���ٴ������Ҳ��Ҫ��MIs������TTCˮƽ����TTC�ǻ���“������¶ʱ�䣨life time exposure��”��ͨ��Ϊ75�꣩�趨�ģ��������ٴ��о��ı�¶ʱ��ͨ��< 30�졣ҵ��ܿ����ʶ����һ�涨ȱ����ѧ�ԣ�������ֽδﵽTTC�Ľ���[5]������ICH M7 (R1)�Ѿ����������ڽ϶̵��ٴ������ж�MIs�Ŀ��ƿ��ʶȷſ�������һѡ�δ��������ã����������ѡ��Ĭ�ϵ�TTC��[6]����Ҳ��ͬ�̶ȵ���Լ����ҩ�з��Ľ��̡�
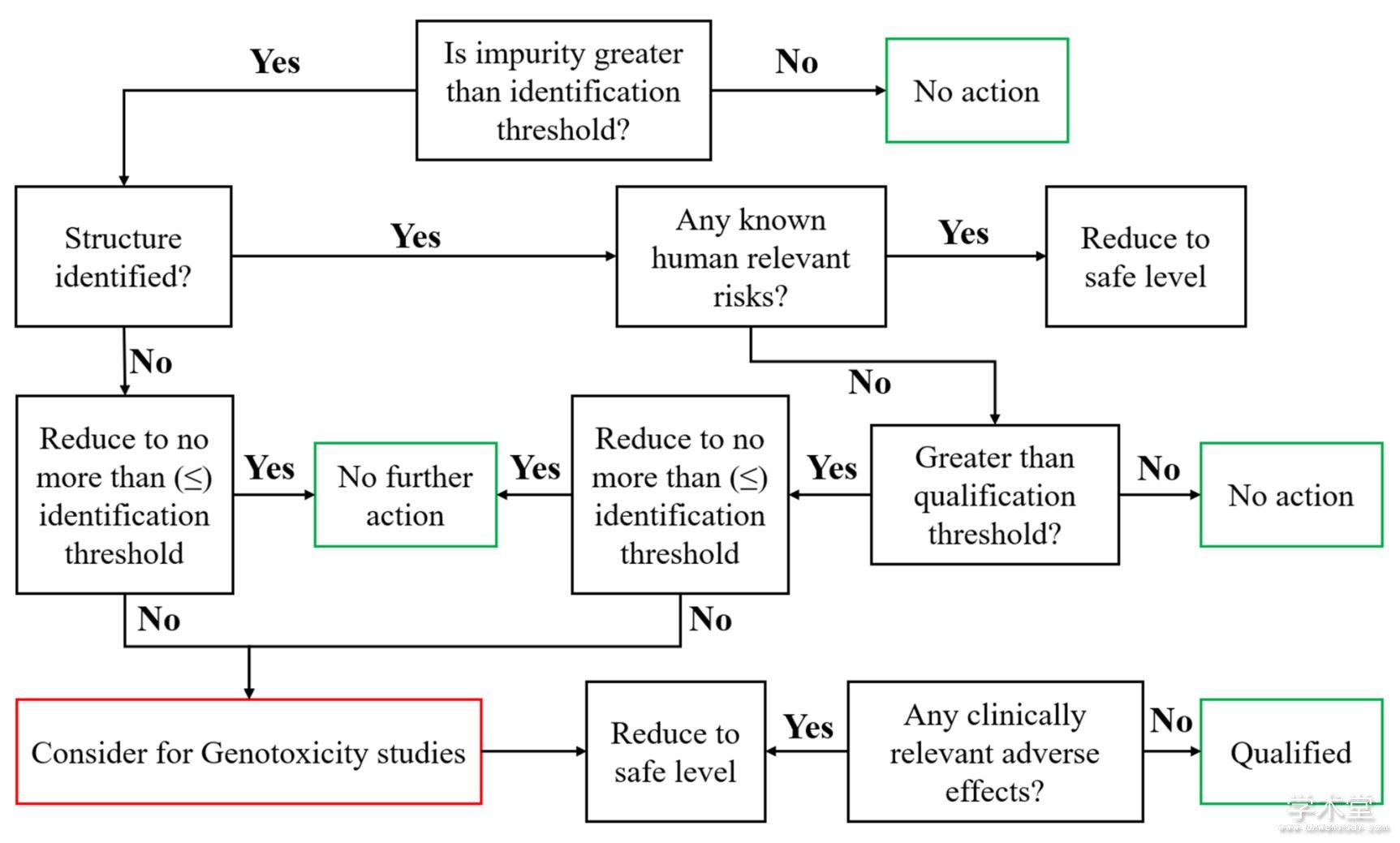
�������ʽ綨��qualification�����������ƵĹؼ����ڡ�����ȱ����Ч�Ľ綨������ͨ�������������������Ӧ�����ܵؿ�������ˮƽ�Է���ICH��Ҫ��Ӧ�ر��ע�������Ƿ������㹻������֤����֪���ʵİ�ȫ�ԡ����ض��������Ƿ���Ҫ���н綨������ȡ���ڻ���ÿ�յ�������������һ���ҩƷ����Ӧ֢����ҩ;��������ʱ��������йء���Ȼ����ҩ�з��ж����ʽ��г�ֵ��о���ҩƷע��Ļ���Ҫ������֪��DZ�����ʵĽ綨Ӧ�ֽν��С�Shaikh��[7]��ȷ�����߰�ȫ�ĽǶȣ��������ҩ�з��н������ʽ綨�ľ�������ͼ1����������������ˮƽ������ICH�ĸ�����ֵ���£��ڶԺ������ڼ��������ʽṹ���м���������������ж�����ܵ��ٴ����գ��۶Ժ������ڽ綨�����ʣ������䵼���ٴ��з���������Ӧ�Ŀ��ܣ��ܶԺ������ڼ��������ʣ�Ӧ����ICH M7��Ҫ����л����Ե�������
����Figure 1 Impurities decision tree for impurity qualification
�����������������������Ŀǰ�ձ鱻������ܲ�����Ϊ�ǻ�������ҩ���е�DZ�ڻ��������ʣ��Ʋ����ںϳɹ��������Ҵ��ȵͼ�������������Ӧ���������Ҫ����ҵ����Բ�Ʒ���Ƿ���ܲ�������Ӧ�����������ȫ����֤��Ȼ��Snodin��[8]��������������ķ�Ӧ���ƺ�ʵ��֤�ݣ���Ϊ�ϳɹ������γɵĻ����������ܴﵽ�������Ŷ���ѧ����ˮƽ��������ѧ�Ƕȣ��������ڴ��е�������Ӧ���ѷ���������ǿ���������²��ܷ���������ת�������Ҵ��ܼ��У�������ҩ������Ħ���Ļ���������̳��Σ�������ֹ�˻��������γɣ���Ȼ�ϳ��и����γ��ȴ����������ȴ��������ױ�������������������ڵ���������ýϻ��������ܶ࣬�������еĶ���ѧ���ݺ�ICH M7(R1)�Ĺ涨�����û������İ�ȫ���������ȴ�����Ҳ�Dz���ѧ�ġ���ˣ���ܲ��ŶԻ�������ع��չ�����ʽ�Ŀ�ѧ��Ӧ���½���������
��������ż��ҩ��(antibody-drug conjugates, ADCs)��Ϊһ�����˵�ҩ�ĿǰICH Q3A��Q3B��Q6B(�����������\����Ʒ/������Ʒ�������������ձ�)����ع涨��������ȫ���������С�������ʵĿ���Ҫ����ҩ���з���������������(The International Consortium for Innovation and Quality in Pharmaceutical Development, IQ)����ר�ŵĹ�����(IWG)�Ը������������[9]�����ڷ��������ķ���������ADCs��С�������ʵķ��������뵰������Ľ�����ԡ�ADC�ĸ�ҩŨ�Ⱥ�ҩ��ʽ�ȣ������ADC��С�������ʵİ�ȫ���������Ժ���ADC����������ϵ�ķ�����ADCs�е�С�������ʲ����Ƿ����뵰�������Ͼ�Ӧ���п��ƣ�ͨ����ADC��Ϲ��յĿ��ƣ��������������嵰�Ľ�ϣ�ͨ���Ժ����������յĿ��ƣ���֤���������ʼ�ҩ�����Чȥ����ʹ֮����ICH Q3A��һ��Ҫ��ͨ��ADCҩ���ȶ��Ե�������Ԥ���Ƽ�������С���������Ƿ�������ICH Q3B��Ҫ�����۽����ʾ��ADC�е�С�������ʺ���ͨ���dz��ͣ��������ᵼ���ٴ���ȫ���ա�
�����Է���ҩ��������Э����ҩ���з�����һ�ȵ㡣2018��10��18������ FDA��ICH����Э��ȫ�����ҩ������[10]�������ȫ�����ҩ������һ���ԣ����ܼලЧ�ʲ����ͼ�ܳɱ�������ȫ�����ҩ�г���ģ��ͨ���������ͷ���ҩ�з��ijɱ�������ʹ�������档2019��2��6�գ�ICH�����˶Դ������˼��[11]����Ϊ��Ȼ����ICHָ�������ڷ���ҩ��������Э��һ�µķ���ҩע���������Ҫ���壬������2019���齨����ҩ�����飨Informal Generic Drug Discussion Group, IGDG����������Խ���������2015��8��18�գ�����Ժӡ���ġ����ڸĸ�ҩƷҽ����е���������ƶȵ����������“��߷���ҩ�������ӿ����ҩ����һ��������”��Ϊ�ҹ��ĸ�ҩƷ���������ƶȵ����Ŀ��֮һ����2016��3��5�չ���Ժ�칫�������ġ����ڿ�չ����ҩ��������Чһ�������۵������Ϊ�ڵ㣬�����������ЧΪĿ��Ŀڷ��Ƽ�һ�������۹������չ��2017��12�£�CDEһ�������۰칫���ַ����ˡ������л�ѧ����ҩ��ע�����һ�������ۼ���Ҫ����������壩�����ҿ���ע�������ҩһ�������۵Ĵ�Ļ�������о�һֱ�Ƿ���ҩһ�����������������ص㡣���ڷ���ҩ��α��Ƽ��������յIJ��죬���ߵ��������ܲ���ȫ��ͬ����Ȼ���·���ҩ��һ��Ҫ�Բα��Ƽ��в����ڵ�“������”ԭ����Ҫ����ICH��Ҫ����п��ƣ��Ը���ҩ���Ѿ����ص�“��֪����”ԭ����Ҫ���м�����Ƚϡ�������һ�������۵Ĺ����Ƽ�ͨ�����ٴ�����Ӧ���˽ϳ�ʱ�䣬�����ҩ�ﲻ����Ӧ��Ϣ������һ���̶��Ͻ�ʾ����ҩ�İ�ȫ��Ϣ����λ��ڷ��տ�������γ��ҹ�����ҩһ���������е���������/���Ʋ��ԣ���ҵ��ͼ�ܲ������ٵ�����ս��
����2�� ��������������չ
���������Ŷ�ԭ��ҩ���Ƽ��и��ֹ�������(��������������)�ͽ��������Ҫ��IJ�����ߣ��Ժ���ˮƽ���ʵı����ͷ�����ҩ������������Խ��Խ�ܵ����ӡ�������������ķ�չ���ر���GC-MS��LC-MS��CE-MS��SFC-MS��LC-NMR��CE-NMR��LC-FTMS�����ü����ķ�չ����ʵ�����߶Ժ�����~0.1%ˮƽ�����ʽ��п��ٷ�����������������֪���ʵĿ���ʶ������Ѿ��൱����[3,12]�������������ߵ�HPLC-MS��/��HPLC-NMR����������Ʒ��������ֱ�ӽ���NMR��������Ϲ���������δ֪���ʺͽ�������ٽ��нṹȷ��Ҳȡ�ýϴ��չ�����������ʣ�MIs��������ҩ���ӳ��ķ������Ҳ�ܵ��߶�����[12]��
����2.1�� MIs����
����Teasdale��[6]��ICH M7ʵʩ����MIs�ķ�����չ������ϵͳ���������µķ���������ע�ض�һ������ǵ�һ��MIs���з���������������Ҫ����и��ߵ������Ⱥ�ר�����⣬��Ӧ�������ٻ���ЧӦ�ĸ��š�����ɫ��(GC)�Ƿ����ӷ���MIs����ѡ��������ЧҺ��ɫ��(HPLC)���ڶԷǻӷ���MIs�ķ���������ͨ���������ȷ�������MIs�Ļӷ��Ժ��ȶ��ԣ����ö��ս�����ʽ������Ч�������ЧӦ��ʹ�ø���������������������ϸ������ϵͳ����MIs��һ������뷽��[13,14]��Sun��[15]����MIs�Ļӷ��ԣ��ӱ�����ʸ��ŵĽǶ������ѡ��MIs���������ľ�������ͼ2����������̽���ȶ���ʵ�������γɻ��������ʵij�������;��������ָ��ҩ����з�������[16]��
����Figure 2 A decision tree for systematic method development for designing methods for analysis of genotoxic impurities[15]
![Figure 2 A decision tree for systematic method development for designing methods for analysis of genotoxic impurities[15]](http://www.xueshut.com/uploads/allimg/190924/36-1Z924155G4Q4.jpg)
����HILIC����ˮ�����ɫ������Ϊ����ɫ�����ر���GC�Ļ������������������GC�Լ���MIs���з�����McCalley[18]��HILIC�ķ�����ƽ�������������ָ�������Ŀ���������HILIC-UV���ⶨ���ण�dalfampridine����5��DZ�ڵķ��㰷��MIs�����У�ɫ������ѡ���ǹؼ���Zorbax�轺����5 μm���ܸ�������ķ������������HILIC������������ZIC-HILIC�������������nitrile-HILIC�����ķ��νϲ���Ӷ��Լ��ɵ��·����Ļ����ʱ���C8��C18ɫ��ϵͳ��ѡ���Խϲ�[18]������ZIC-pHILIC��ˮ����ɫ����������CAD��NQAD��������ں�TFA�������пɶ�12�ֲ����������յļ���MIs������֪�°����£�hydrazine�����вⶨ[19]��Denton��[20]����HILICʵ���˶����ȱ���ȩ��2-chloromalonaldehyde���ķ�����Dou?a��[21]����HILIC-MS��������������͡��vortioxetine�������Ķ�(2-���һ�)��[2-chloro-N -(2-chloroethyl)ethanamine]���������������Խ�һ�����MIs�����������ȡ�Grinberg���������������Ϊ�������Լ��������������ʼԭ���е����������(dimethyl sulfate, DMS)������������������������������N-����ण�����HILIC-ESI-MS��SIM���ģʽ��DMS�����Է�ΧΪ0.05~10 ppm��LOD ��LOQ�ֱ�Ϊ0.4��1 ppm[22]���������͵��������Լ�BPPC [butyl-1-(pyridine-4-yl) piperidine-4-carboxylate]������HILIC-MS/MS����������ΪAPI�����±�������������εij������������ɸ�鷽����ǰ�ߵļ��ˮƽΪ0.1 ppm������Ϊ1 ppm���ҷ��������ܻ��ʵĸ���[23]��
��������ɫ����Ȼ����ΪGC-MS��HPLC-MS�ĸ������������ڷ���ǿ���Ե�MIs������Ȼ��alkyl chlorides�����µȣ����������Ľ�չ��С[6]��Frenzel��[24]������ɫ�����г��õ�Ĥ���������������������������߹�����ȡ�������ⶨ������ɳ�����еĺ����ǰ�����Ʒ��Һ�еļ�����ɳ������������IonPac CG12A������ȡ���ϣ��ǰ�����ɫ�ײⶨ��Ԫ��CG12A��������CS12A��������������������������LOD��LOQ�ֱ�Ϊ0.02��0.04 μg·mL-1 [25]�����ٽ�ɫ�ף�SFC��Ҳ����ΪHPLC�Ļ���������Lesellier��Westd�Խ�����SFC�ļ�����չ����������[26]���Գ��ٽ������̼Ϊ�����࣬�״�Ϊ���Ը��Լ����������ֲ�ͬ�ı�����(Synergi polar RP��Cosmosil 5PBB)���Ƚ�SFC�Զ����(PAHs)�����ѡ���ԣ�����PAHs��SFC��HPLC�еı�����Ϊ��ͬ�����Լ��״���Ũ�ȶ�MIs��Synergi polar RP���ķ���Ӱ��ϴ���Cosmosil���ķ���Ӱ���С[27]���Ƚ�SFC-ELSD��HPLC-ELSD����PVC�����е��ܻ�����ATBC��DEHA��DEHT��TOTM����SFC-ELSD�������ȸ��ߣ���HPLC-ELSD�ľ����Ը���[28]��ëϸ�ܵ�Ӿ(CE)����ɫ�����Լ�����Ʒ�������õķ���ѡ���ԣ���Ȼ�������Ƚϵͣ�ͨ����������MIs��������CE���������ߣ����������ļ���ɴ�2 ~ 3 ppm����ΪHPLC�Ļ������뼼�����ڷ���ԭ��ҩ�л��������/���������������������������������PMIs�о���Ӧ��[6]��
�������ἰ���������Ϊ�µ�MIs����Ҫ��һЩ��Чż����Ӧ����ľ-����(Suzuki-Miyaura) ��Ӧ�в���������������MIs����ͨ������Ԫ�صIJⶨ����ͨ����ѧ������ϵ�õ��京������ˣ�����ICP-MS����ж���Ʒ�еIJ�������вⶨ�����������ȸߣ�LOQΪ0.8 ppm����ֵΪ40 ppm����ѡ���Ժã��ҿɱ��ⳣ���Ļ��ʸ��š�Patel��[29]�Ը÷����IJ������ü��Ż��г�����������������������黯����MIs�IJⶨ������GC-MS ��HPLC-MS����������������������������ICP�����±��Ԫ�صIJⶨ�Ѵﵽppb������Ϊ�黯���ķ����ṩ���µĽ������[30]������HPLC-ICP-MS�����������4-��-1-����������3-�ⱽ�����Լ�������������������LOD��LOQ�ֱ�Ϊ0.2��0.5 ppm�����Է�Χ(μg·g-1 API)Ϊ0.5~50 ppm��1~50 ppm��ȷ��Ϊ95.1% ~ ?114.7%���ظ��ԣ�RSD��Ϊ6.2%[31]��������ͬ�ķ����ⶨ�黯�����¿ɵõ����ƵĽ���������������������Լ�������߷����������ȣ�LOD��LOQ�ֱ�Ϊ0.06��0.2 ppm[32]��
����2.2�� Ԫ�����ʷ���
��������Ԫ�����ʿ�����ҩƷ����������б����������ղ�Ʒ�Ի������Σ����ICH Q3D�����ҩƷ�е�Ԫ������Ӧ���ж��ԺͶ������ƣ�ҩƷ�и���Ԫ�����ʵĿɽ��ܵ�ÿ�սӴ���(permissible daily exposure, PDE)�����ҩ;���йأ�USP���°䲼��ͨ��<232>�ж�ҩƷ�е�24��Ԫ�������������ȷ���ʿ�Ҫ��1����Ŀǰ����ҩ���з�����ط�������Ҳ������ICH��£��
����Table 1 The elemental impurities in pharmaceutical products e parallel ICH Q3D and USP general chapter <232> guidelines for permissible daily exposure (PDE) limits depending on different routes of administration, i.e., oral (O), parenteral (P) and inhalational (I)[33]. aThe elements that need to be considered as part of risk assessment if not intentionally added
![Table 1 The elemental impurities in pharmaceutical products e parallel ICH Q3D and USP general chapter <232> guidelines for permissible daily exposure (PDE) limits depending on different routes of administration, i.e., oral (O), parenteral (P) and inhalational (I)[33]. aThe elements that need to be considered as part of risk assessment if not intentionally added](http://www.xueshut.com/uploads/allimg/190924/36-1Z924155615C8.png)
����ͨ��ҩƷ�ղ�Ʒ�вд�ĺ���������Ir��Os��Pd��Pt��Rh��Ru��Ԫ�����ʣ�������ԭ�Ϻϳ���ʹ�õĴ����йأ�Cd��Hg��Ni��Pb��Ԫ�����ʿ���ͨ�������е�ˮ���ܼ����ϳ��Լ������ϣ��ȶ�����������ճ�ϼ������ϡ����Ϻ�Ϳ�ϣ���;����Ⱦҩ���Cr��Cu��Mo��Ni��V��Ԫ�����ʿ�������������в�Ʒ���Ϲޡ�������������ߡ���װ�����ȱ���ĽӴ���Ⱦ��Ʒ[33]��Jenke��[34]����ҩƷ�����Ͱ�װ��������Ԫ�����ʵķ��ս�������������ҩ��Ӵ��ĸ��������ߡ���װ�����Ȳ��ϣ���ҩƷ�Ӵ�ʱͨ��ֻ������Ԫ��ʵ���ת����ҩƷ�С���ˣ���ȻijЩԪ����������Ȼ�������ձ���ڣ�������ҩ���̺Ͱ�װ��������ҩƷ�ķ��ղ����ߡ�Boetzel��[35]������һ������ҩ��˾���������İ���201�ָ��ϡ�26 723��������ݵ�Ԫ���������ݿ⣬��Ŀǰͬ�����ݿ��ٮٮ�ߣ�������Ѹ����������ҩƷ�ķ���������Paskiet��[36]��ע����ӳ��õ��ܷ���ʵ������Ԫ�����ʵķ��ս�����������
���������ϵ�������-����(ICP-MS)��Ŀǰ�ⶨԪ�����ʵ�����ֶΣ�����ǵ����ϵ�������ԭ�ӷ������(ICP-AES)�����Ǿ����ڶ�����Ʒ������ͬʱ������Ԫ�����ʣ�������һ�ֻ����ض�Ԫ��������Hg,��As��Cr���вⶨʱ����ͳ��ԭ�����չ���(AAS)Ҳ�ɵõ�����ķ������[33]��Wollein��[37]����ICP-MS�������ϵ�������-�������(ICP-OES)��ԭ�����չ���(GFAAS, CVAAS, HGAAS)�����г���113����Ʒ��21�ֽ������ʽ����˷�����Li��[38]������31��190��ҩ�ø�����ƷԪ�����ʵļ�ⷽ�����ⶨ�����ICP-MS�ر������ڶ����������ߵ�ע����������Ƽ�����Ԫ�����ʵIJⶨ���������ܶȣ�RSD��С��4.5%��ICP-OES�IJ����ϼ��Ҿ��нϿ�ķ����ٶȣ�ͨ��������ԭ���ϺͿڷ��Ƽ��е�Ԫ�����ʷ��������ַ��������IJⶨ�����������USP ͨ��<232>��Ҫ��[33,37]��Menoutis��[39]���ó�������(UN)��������ϵ�����ԭ�ӷ��������ICP-AES���ⶨ����һ��Ͷ���Ԫ�����ʣ��ϳ���ICP-MS�������и��͵ļ���ޡ�Balaram[40]�Ը���������������������Яʽ������ԭ�����չ���(AAS)��X-����ӫ�������(XRF)�����������ӻ����(INAA)����ICP-AES��ICP-MS��Ӧ�ý�����������
������������������(MWAD)��������Ʒ����ǰ������Ԫ�����ʷ����Ĺؼ����ڡ�USP ͨ��< 233 >����������ͨ�÷������ֱ�����ICP-AES������ICP-MS������Jin[41]��Ԫ�����ʷ�������ǰ�����еij�����Ⱦ;����������������ʵ���һ������Լ�������Ŀ����DZ�֤�ⶨ���ȷ�Ĺؼ���Muller��[42]ͨ���Ƚ�Ũ���ᡢ��ˮ��aqua regia��������ˮ��inverse aqua regia����4��ԭ��ҩ��������Ч�ʣ��������������ˮ�����룬ǰ�߿���Ч����500 mg��Ʒ������������250 mg������Ʒ�����⣬���еIJⶨԪ��(��OsԪ�����γ�OsO4Ӱ�������)���нϺõĻ�����(91% ~ 109%)��da Silva ��[43]��������ˮ�����������ڲ���ICP-OES ��ICP-MS���ٷ���As��Cd��Hg��Pb �ļ����������ⷽ����Paskiet��[36]�Ƚ��˶��ֶԺϳ�����Ԫ�����ʵ���ȡ������
����2.3�� ������/��ȡ�����
����ҩƷ�еĽ�����һ����Ϊ���ڳ������������£�����ҩ��Ӵ���ʵ�壨��װ���ϡ�ע��������Һ�ܵȣ���Ǩ����ҩƷ�еĻ�ѧ���ʣ���ҩƷ�и��������ķ�����������������һ���֡�����ȡ��һ����Ϊ����ʵ�����ܿ���ȡ�о��д�����Ʒ�ͷŵ���ȡ�����еĻ�ѧ���ʡ���ȡ���������ӷ��ԡ���ӷ��ԡ��ǻӷ��Ե��л������������չ�ܿ���ȡ�о���ϣ���˽�ҩƷ��ʵ�Ľ������ף������������������İ�ȫ����[44]�����������ǰ�������Դ����ƣ�QbD�����������ҩƷ����ɡ���װϵͳ���������״����ҩ��ĽӴ�����ȣ������õ����������������ҩƷ�н�����İ�ȫ�ռ䣻Jenke�����ϰ�װ��ע��ҺΪ������֤�˸�����Ŀ�����[45]��
������ҩ�þۺ��������ȡ���м�������540���ֻ�����Ķ���ѧ��Ϣ����δ�۲쵽���õ�ˮƽ(no observed effect levels, NOELs)��δ�۲쵽�к����õ�ˮƽ(no observed adverse effect levels, NOAELs)������������ж�����(lowest published toxic dose, TDLOs)�ȶ���ѧ�յ�ָ���ѱ����ܣ������ڽ�����ķ�������[46]����һ����Ӫ����������-��Ʒ�����о���(PQRI)�����Ľ�����/��ȡ�﹤��С�飬��ҩƷ�з��������������/��ȡ���йصĿ�ѧ�������������̽�֣�Ŀǰ���ڽ�����/��ȡ������ı�����������Ͱ�ȫ��ֵ�ȷ����ɹ�ʶ���ҷֱ����ڶԿ�ǻ���������ǻ�Ƽ���ע����������Ƽ��еĽ�����ķ�������[47,48]������ˮ���л���ȡ�ܼ�������ע�����װ����21�ֳ��þ۱�ϩ��֬������ȡ�о�����������ȡ�����������ע������ڡ����������ʱ��DZ�ڷ���[49]����Ȼ���ڴ������յIJ��죬�ڲ�ͬ��ȡ�����²�ͬ�ľ۱�ϩ��֬���л���ȡ���ײ���ϴ������ض�����ȡ�ɷ��翹�����ȿɽ����Ϊ��ͬ���飬ͬ����Ʒ����ȡ�����������ԣ��������ȡҺ��Ԫ�����ʣ������衢������ͼ����������������ϵ͡�
����������/��ȡ���������һ�ؼ�������α�֤���н���/��ȡ�������ʶ��õ������ɫ���dz��õķ���������Jenke��[50]���û�����ɫ��ϵͳ��GC��HPLC��϶��ּ�ⷽʽ������ҩ���װ����������ҩϵͳ�ȳ��õ����ϲ����е���ȡ���ף�Ϊ��ֹɫ��������©��ijЩ��ȡ��������л�̼����(TOC)�ⶨ�������Ը���ˮ�����������Һ��ȡ���������������ԣ����ڶ�������ˮ����ȵķ����еõ��˽Ϻõ�Ӧ�á�����ȡ���в��ʺ���GC���з����ķǻӷ��Լ��Ȳ��ȶ���������ɴ�����ѧ�������������������ɵ�UPLCϵͳ�У�22 min����ʵ�ֶԶ��ֳ�����ȡ���еĴ����Ի�����������г�ֵķ������⣬�ڶ�ʵ����ȡ��Ʒ���з���ʱ����ʹ���������Dz����ݵ��л���ȡҺ��Ҳ�����ɫ��ϵͳ��������Ӱ��[51]��
����ͨ��ģ�����(Ǩ��)���飨�ɵ��ܶȾ���ϩƿ���۱�ϩƿ�Ǻͽ�������ɡ���ȡ�ܼ��ֱ�ΪpH 2.5�Ļ�����Һ��pH9.5�Ļ�����Һ�������1:1�������/ˮ������ʾע����͵��ۼ����ĵĽ������ס����ĵ���ȡ��������ĵĻ�ѧ�����ṹ������أ�������ȡ���ʺ;�������ﻯѧ���ʵ�Ӱ�죻��Ȼҩ�������ֱ�ӽӴ����ܼ���ijЩ�������Ǩ�ƣ��������Ƿ���Ǩ�ƺͽ������Ⱦ�����[52]�������е���������ͷ�߾�������ã��������������ҩƷ�ܽ�ʱ������ǹ���ͻ������������[53]������GC-MS�����Ľ����ӷ��Գɷַ������ݿ⣬�������Կ��ٷ������ý����е���Ҫ�ӷ���Ǩ��������Կ���ȷ������ͷ�߾�������õ�Ǩ������ģ���������飬���γ���Ч��ͷ�߾���-����������������ԣ���������Եؽ��������ʿط���[54]��
����2.4�� AQbD���Ӧ��
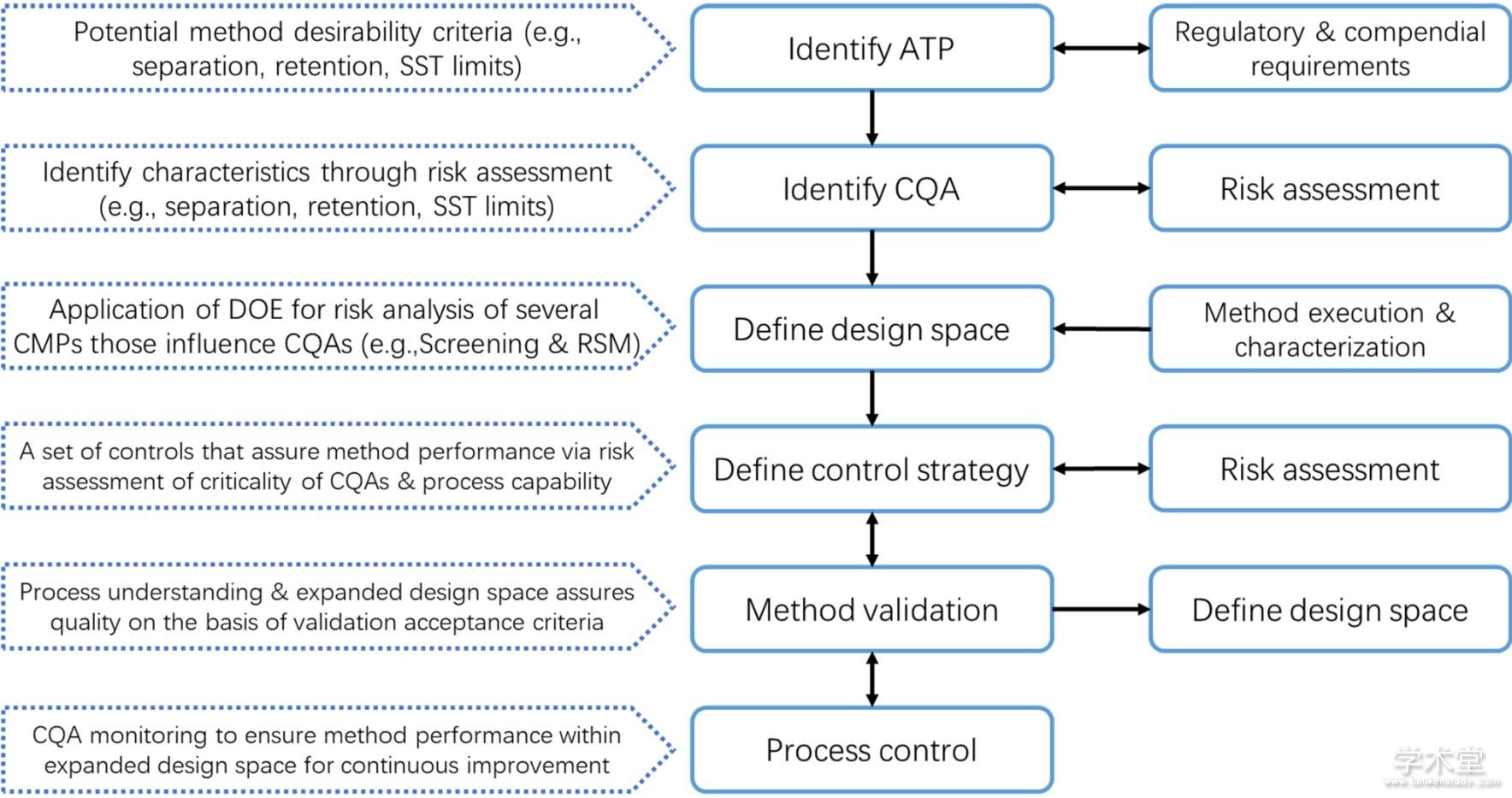
�������չ���������������֤��ϵ�Ľ��ܽ���DZ�֤ҩƷ��������Ҫ���ڡ��������ƹ����У���֤�����ʿط�������������ҩƷ�������ڶ��������õ�ר���Ժ������ԡ�������������Դ����ƣ�analytical quality by design, AQbD��������Ϊһ��������չ����ķ����ۣ��������ڽ���������������ʱ���㷺���ܡ��봫ͳ������Դ�ڼ�⣨quality by testing, QbT��������Ƚϣ�����ݷ���Ŀ��ı仯��Χ��analytical target profile, ATP��������ʵ����ƣ�DoE���ķ�����ͬʱ���ǹ�������������ķ�����ȷ����ƿռ䣨design space, DS���������̶ȵı�֤��������Ч��[55,56]������AQbD������ķ�����������������ʵ������ڲ����������(method operable design region, MODR)�ڱ仯���������и��õĴַ��ԣ��ɼ���ʵ���еij������ƽ����out of trend, OOT���ͳ����������out of specification, OOS��[57]��ͬʱ�����ڿ�����ȵؼ����ڷ���ת�ƣ�transfer��������ȷ�ϣ�verification���ͷ�������еĹ������������ڽ��ͶԷ��������������ڹ�������������ơ����������ͷ�����֤�������ļ춨�������ķ���������֤�ͷ���ת�ƣ��ijɱ�[58]��
����Dispas��[56]�Խ�����AQbD�����������е�Ӧ�ý�չ�������������ڻ���AQbD����� HPLC���������У���ѧ����ѧ������ȷ�����˵���Ʒ���ɸѡ�ؼ�Ӱ�����ӣ�����������ϵģ��ȷ�������IJ����ռ�ȷ��棬������Խ��Խ��Ҫ�����ã�ͼ3��[59]��Tumpa��[60]����˻���QbD�������HILIC������ָ��ԭ��Zhang��[61]���û�Ϲ��̱�����mixture-process variable, MPV����ƣ������˻���QbD�����HPLC˫�ݶ�ϴ�ѷ����������������������ȶ��������н������ʵIJⶨ��Taheri��[62]�ڶ�����������celecoxib����ϴ����İ��Ʊ������У��������ĸ�����Ƴɹ���ѡ������ɫ����������
����Figure 3 Key steps in QbD driven chromatographic method development
����������HPLC-MS/MS�����ü��������Ŀ�ͬʱ�ⶨ��ʮ��������Ŀ�������ķ�����������Բ���ũҩ�ȵIJⶨ������ͳ�ķ�����֤Ҫ��ͨ����Ҫ���ñ����뷨�Զ���ȷ�ԵȲ���������֤������֤������ʱ���������ҵ�Ŀ�������仯���������µĿ��ƶ���ʱ�������¶Է���������֤��ʵ���������Ʒ�ļ����������ˣ������ܵõ�����ȷ����֤�����Alladio��[63]���û�ѧ����ѧ�����������˶���Ŀ�������ı���ʱ�䡢����ЧӦ�������ʡ�LOD��LOQ����Ԥ���ƫ��С���˷�ģ�ͣ������������з�������Ŀ�������ķ�����������ȡ����Ԥ�ڵ�����Ч�������ö����ṹ-������ϵ(QSRR)��������Ԥ�������ı���ʱ�䣬�������ڶ��������Ƶ��˽⡣��δ֪��ֱ���ֵ��Ԥ�������ѡ��������������ٷ���������ʱ�䡣Amos��[64]�Խ���QSRRģ�͵ķ������ؼ����Ԥ�⾫�ȵȽ������������������ʵ�ɫ�����ƶ�ָ����chromatographic similarity index�������ֲ�QSRRģ�Ϳ�ȷԤ�����ʵı���ֵ[65]���Ƚ�4�ֳ��õ�����QSRRģ��Ԥ��ȷ�Եı�����������ΪԤ����������ٷֱ�(RMSEP)�Ƕ�QSRRģ��Ԥ����������ѹ���ֵ[66]�����ö����ṹ-������ϵ(QSRR)ģ�ͣ�ͨ��Ԥ���������5�ֲ�ͬHILIC�̶����ϵı���ֵ��������ѡ�������˷���Ĺ̶���[67]�����ö����ṹ-������ϵ(QSRR)�����ˮ����ģ��(HSM)��Ԥ��������ڷ���Һ��ɫ��ϵͳ(RPLC)�еı���ֵ������Ԥ��ҩ�������Ƿ���ҩ����Գɷ֣�API���Ĺ�ϴ��[68]����ȱ�����ʶ���Ʒ�����ж�һ���½�����ɫ�����Ƿ���Զ�ҩ�������е���֪���ʶ��ܼ��ʱ������QSRRģ�ͣ�ͨ��Ԥ����֪���ʵı���ʱ�䣬�������жϷ�������֪���ʵļ������[69]��������Ԥ�������ʵ�ɫ����Ϊ[70]��
����2.5�� ����
����ѡ�����˵�ɫ������Ȼ��ҩ�������������ȵ㡣��ͬɫ������ѡ���Բ��죬��������������������ҩ�������صij�������ʷ�����������ˣ���ο���Ѱ�ҵ��������˵�ɫ��������Ϊʵ��Ĺؼ���������ˮ����ģ�͵�ɫ����������ϵ����ɫ������ѡ�����ѡ�����ƻ���ɫ������ѡ����ѷ���ɫ�����й㷺Ӧ��[71]�����ƶ���ɫ����ѡ��ģʽ�ķ�չ[72,73,74]��������ˮ����ģ�ͣ�����Ӱ��β-������������ѷ������ʶԷ���Ĺؼ�ɫ����������������A��������˲���A���������[75]���Կ���ù�����ʷ���Ϊ����ѡ�������ڷ����ѷ������ʶԵ�ɫ�����������Ӧ����ˮ����ģ������ض�������ѡ�����ɫ�����IJ���[76]�������о�������ɫ����ѡ��������ʵ�û�����ȡ�õĽ�չ����ͨ������ģ�ⷽ������Ӷ���ѧ�����ؿ���������ؼ���ʵ��ɸѡ������ɫ����ѡ��Ӧ���о���Ŀ��[77]��
����ҩ�����������е���һ�ؼ����Ƕ����ʵļ���붨�����ڶԸ������UV���յ���Ʒ�ķ����У��������ܽ���aerosol-based�����ĸ���ͨ���Լ�����ر��ǵ������������charged aerosol detectors��CAD���͵绯ѧ�����������Խ��Խ��Ҫ�����á���ȻCAD��������ձ���Ϊ��һ�������ͼ������������û�ж���Ʒ�������ʵ�ֶ����ʾ��Ժ�����ȷ���ƣ�Ȼ����50�־��й㷺������ѧ���ʵĻ������HPLC-CAD�ⶨ�������������ڴ�ͳ������У������������ͨ�����Ƽ���������Ա��������У�����ɵõ���ȷ�Ķ���������ر��ǿ���������߶��ܶȽϸ����ʶ�����ȷ�ԣ���NMR������ƽ�����Ϊ5.8%��[78]��
�����������������������ʶ���Ʒ���ж����붨��������������ķ�������ҩ����������У����ʶ���Ʒ��ʹ��ҲԽ��Խ�ձ顣���ͬʱ�������ʶ���Ʒ���Ʊ�����Ӧ��ȫ�������ڵĹ���Ҫ��ҲԽ��Խ�ϸ�[79]���ҹ���2016��5��ʩ�еġ���ѧҩƷ��ע������걨����Ҫ�����У����У��Բ�ͬע������ҩƷ����ԭ�Ϻ��Ƽ����ƹ�����ʹ�õĶ���Ʒ���������ɷֶ���Ʒ�����ʶ���Ʒ�����ṩ������ϡ�����ҩ�����Ʒ���������ṩ���š����ȡ�˵�������ϸ��Ϣ����Ҫ�ṩ��Դ֤�������ƹ��������ʹ���������Ʒ�����ƶ���Ʒ�������ṩ��Դ֤���⣬�����ṩ�ṹȷ֤���������Լ������궨���̵���Ϣ�����Э��������ܲ��Ƕ����ʶ���Ʒ��Ҫ����߸������ʶ���Ʒ�Ŀɼ������������о�����һ�ؼ���[80]�����û�����ʶ���Ʒ���ж���[80,81]�����ü�У�����ӵ����ɷ��������շ��������ǽ������֮һ�������������ɷֵ�У��������0.9~1.1����ֱ�Ӳ������ɷ��������շ����㺬���������������ɷֵ�У���������ϴ�ʱ������У����������������Ӧֵ����Բⶨ�����Ӱ�졣��������ƽ��ԭ��������HPLC-DAD���ELSD��MS���������Ա�֤У�����Ӳⶨ��ȷ��[82]��Ȼ�������ö���NMR���HPLC����������У�����Ӳⶨ�������ʶ���Ʒ������ȷ��������������Ч;��[83]��
����3�� ���ʷ�������Ӧ�ý�չ
����ҩ���е����ʰ����������ʺͽ������ʣ�����ҩ��Ľṹ����Դ�����յ��ص㣬��������˼��һ��ҩ��������������ԣ����������Ƶ�һ����Ծ���Բ�Ʒ���������ƾ��и�ʵ�ʵ����塣�������[84]�Զ��������ҩ���������Դ����ⷽ�����������Լ����۱���������ݽ�����������Ԭҫ����[85]�ع˰���������������/���ʷ��������ķ�չ���̣������˳�������������������/���ʷ�����������ȱ�㣬��������Ӧ��Һ��ɫ�����尲��绯ѧ��ⷽ���ڰ��������������ַ��������ƣ�̽�ָ���ҩ��δ���ʿصķ�չ����
����3.1�� ������������
���������ȷҩƷ�е�������ɽ����������˵ķ�����������������������Ҫ��������⡣�ڹ��ڸ����ض�Ʒ����ҩ��������صľ������ʽṹ��������ױ�����������������������������иĽ����Ż�����Ȼ���нϺõ�ʵ���ԡ������������[86]���������ǿ���[87]���˾���[88]ԭ��ҩ����ù�����[89]�������������淥��͡��Ƭ[90]�й�����HPLC���������ĸĽ��ȡ�������ԭ�Ϻϳɼ��Ƽ����յ��ص㣬�������˵������������������������ս���������������Ϊ�µ��ȵ㡣�������ƥ[91]����������ŵ����[92]����������[93]ԭ�ϼ����ᷥ���Ƿǿ�ǻ����Ƭ[94]�й����ʵķ����ȡ�
����ҩ���ȶ���������Ϊȷ��ҩ���н������ʵ���Ҫ���ڣ�ǿ�ƽ���������ΪԤ��ҩ�ﳤ���ȶ��Ե���Ҫ���ߣ������˽�ҩƷ�Ŀ��ܽ���;���ͽ������ȣ�����ָ������������������[95]��ͨ�����ȶ������鷽���ĺ�����ƣ�������������ҩ�ﴦ���ĺ����ԣ�ȷ����Ʒ����Ч�ڡ������ں����������ȣ������ܵ���Ҫ�����ȶ��������в����Ľ������ʻ�����������֤����������������Ч��[96]����ͨ���������ȶ���ʵ����3����δ֪�������ʵļ������������µ����ʿ��Ʒ���[97]������ǿ�ƽ������黹�����������������ж����ʵĽṹ����ȷ�ϣ�������ǿ�ƽ����������������Ƭ[98]��ͷ��������ˮ����[99]����ɼ������ע��Һ[100]�������ȡ����⣬ͨ���Բ�ͬ����������ע����ͷ�����������������ʽ���;����ͬ�ķ����������ǿ���ƻ��������Ʒ����Ҫ���ʵ���Դ�ͽ���;�������о������Ը�ȫ����˽��Ʒ�����ԣ���������ҩƷ������[101]��
�����ڹ��������Գ����У�ͨ�����г��в�ͬ��ҵ��ͬ��Ʒ��������ϵͳ�Ƚϣ����ԽϺõ�����ͬƷ��ҩƷ�Ƽ�����������ɵ���Ϣ�������������������������������Զ�����ҩ����������ԭ�ϼ����Ƽ�[102]����ϳɿ��������к�ù��Ƭ[103]��������ù�ؿ�����[104]����ѧҩ�ڷ��Ƽ��շ���͡�Ƽ����Ƽ�[105]����ɳ̹����[106]�������忹��ҩͪ���ᰱ������ע��Һ[107]��˳��ע���[108]�������������[109]���й����ʷ��������Ľ����������������������Ը��ӳɷֵ���Ʒ����ύɳù�ؽ��з����Ż����ɻ�ø�����ķ�����[110]�����Բ�ͬ��ҵ��Ʒ�����ıȽϣ����������۲�Ʒ�����IJ��죬��ʾ��������������������ԡ����������������[111]��ע���û���������[112]����ע����ͷ��������[113]�ȵ��������ۡ��������������������������������Ը��õ��������������������Ĺ�ϵ������ʵ�־��ضԹؼ��������յĿ��ơ��簢Ī���ֿ���ά���Ƭ���������������ʾ���Ƽ������а�Ī������ˮ�������ˮ�밢Ī���ֱջ�������IJ����������[114]��ͷ�����������������������ʾ��������ǰ��ʪ�ȵĿ����ǹ��տ��ƵĹؼ�[115]��ע����������ȷ����������������ʾ������AΪˮ�����ʣ���Ҫ���Ƽ��䶳����������룬����B ������CΪ�������ʣ���Ҫ��ԭ��ҩ�ϳɹ������룬�빤����ʹ�õĵͼ������й�[116]����ע���ð������������������������IJ�����Ҫ�백��������ԭ�ϵ����������йأ��䶳���﹤�յİ���������ԭ�Ͻ���ý�ᾧ���ղ�Ʒ�����ȶ����Ƽ��������¶ȵȻ������ص�Ӱ��[117]��
����ѡ�����˵�����ɫ��������HPLC��������ʵ�ʲ�Ʒ�е��������ʽ��з�������Ȼ��ҩƷ�������Ƶij��÷���������ù轺����Ϳ��ֱ������-����3��5-���ױ���������������Ϊ����������ɫ������������������칹������[118]���������з�����ԭ�ϼ�Ƭ���ж�ӳ�칹������[179]�IJⶨ�ȡ�Ȼ�����ó��ٽ�����ɫ����ҩ���ж�ӳ�칹��ķ����ǽ��������о��ȵ㡣���1,4-����������������������ƽ�������ƽ��������ƽ�����۵�ƽ�����������ƽ�������Բ��[120]������������R-��ӳ������Բ��[121]�ȡ�
�����Զ�����ҩ������ʿ����о�Ҳ�ܵ���ע�����Զ��ĺϳ��е��м��塢DZ�ڸ�����ͽ�������Ϊ�о������������������֣�һ�ֺϳɵ����Ծ��ģ��й����ʷ�������[122]������LC-MS�����ٽ�����ע��Һ�еĽ�������[123]����������ɫ��������ҩ���������Ǻ����ķ���[124]�ȡ�
�����������ࡢ���������Ƚṹ��Լ��Ҳ�����UV���յ�ҩ��/���ϵ����ʷ����о��Ľ�չ�ϻ�������Ȼ�в��ó��ٽ�����ɫ���������ü������Է�����/�����ṹ�����������[125]�ı���������Ҫ�о��Բ���HPLC����ĩ�˼�⣬��ⶨ�Ŷ������þע��Һ���Ŷ�������й�����[126]�����ᰱ�������ǵ��й�����[127]�ȣ��������·�����Ƶ�ɫ��ϵͳ��HILIC�ȣ�������͵ļ������CAD��NQAD�ȣ������ٽ�������ķ�չ��
������β-�������������оۺ���ķ�����Ŀǰ�й�ҩ�����ص�Sephadex-G10����ɫ��ϵͳ�Ѿ��������ִ��ʿص���Ҫ�������ڸ�Ч����ɫ����������ר����ҲԽ��Խ�ܵ���ս[128]�����ڶ�άɫ�����ķ�չ���������л��������Է���ؽ�����ɫ��ϵͳ�е�ɫ���ڳ���HPLCɫ��ϵͳ�н��ж�λ�����LC-MS�������������ʷ��������н��ۺ���������Ϊ�ض����ʽ��п���[128,129]����Ҳ����Ϊ����β-�����������ؾۺ���ķ�չ����
����3.2�� �����ʽṹ����
��������LC-MS�����Բ�Ʒ�еĹ������ʺͽ����������ɷֵĽṹ�������з���,��Ȼ��Ŀǰʶ�����ʽṹ����Ҫ����[130]����ͬϵ��ҩ�P�����ѽ���ɵ�̽�������ڶ����ʵ�ʶ�����ϵ�[131]��7��β-���弤�����ĵ����������ѽ���ɽ�����̽�֣�Wang��[132]��β-�����������غ��俪����������ѽ��������˱Ƚϣ�Qian��[133]��ͷ�߿�������3�칹��������ѽ��������˱Ƚϣ���NMR���������ü��������ʶ��Է����е�Ӧ��ҲԽ��Խ�㷺[134]��
�������ڽ������ڶԽṹ�����Ҵ����������ĵ�δ֪���ʽ��м���ʱ���ձ�����Ʊ����룬�����ø��ֲ���������ṹ�����Ʋ�IJ��ԡ���������������[135]����������[136]���ʵȵļ��𡣶����ø߷ֱ����ף�����һ�������������ѽ�����Ʋ⻯ѧҩƷ�����ʵĽṹ����Ϊ����������������[137]���⺣�����й�����O-����������N-����������ȷ�������ͽṹ�Ʋ�[138]�����õ��������ӻ�����2������ģʽ������ȼ�Ⱥ���м���[139]�����淥��͡�м���[140]�е����ʣ����÷���ʱ�������Ʋ���ƥ��¡[141]���������������Ƭ[142]�е����ʽṹ�����ø߷ֱ����ʱ����������ɳ̹��[143]�ʹ���ҩ�ﰬ��Ī��[144]�е�δ֪���ʡ���άҺ��ɫ��-�������ü���������ɫ�����������ֱ�����������������⡣�����ڷ�����������Ƭ��������[145]�Ͷ��������ע��Һ�����ʽṹ�Ľ���[146]�ȡ�
����3.3 ����/�������ʷ���
���������������ʺ�MIs�ķ����������ķ�չ�Ͽ졣�����õ�[147]����UPLC-MS/MS�������ⶨ�����������ԭ�ϼ�ע��Һ�е���������1-��-4-����-1, 2, 3, 6-������ण������ɵ�[148]��������ɫ�װ��෨���ά����B6��2-��������е��軯����õ�[149]�����Ҵ�������������ɫ���ⶨ�������ԭ��ҩ������ȩ�ͼ���ĺ���������HPLC��ǰ���������ⶨ��ɽ�����ศ����ȩ������[150]��
������MIs�ļ���ǵ�ǰ���ڵ��ȵ㡣��������[151]��MIs��ҽҩ��ҵ�еĿ�����Դ�Ƚ����˷�����������[152]��ҩ���з���MIs�Ŀ��Ʋ����뷽����������������ѩޱ��[153]������ҩ���л�������MIs���о���չ��л���ǵ�[154]��MIs����������ǰ�����������о���չ��������������Ȼ����HPLC-UV������ʱ������MIs�����ʿص�Ҫ���������ɳ��[155]���䱽����[156]�б�����MIs�ķ����ȣ���Ϊ��߷�������������ȷ�ԣ�������Dz���HPLC-MS��HPLC-MS/MS��������Ծ�ʯ�ᷥ�����[157]�ͷ������[158]�к���������MIs�IJⶨ������������������[159]�Ͳ���������̪����[160]�мױ���������MIs�IJⶨ; ���������а�����������MIs�IJⶨ[161]������ɳ���е�3-������-1-[4-(5-����������)����]-5,6-�������-2(1H)-ͪ (2)[162]�����������е� (2S)-2-����-4-��-1-[(2R)-2-�����������]-1-��ͪ (3)[163]��MIs�IJⶨ��Ȼ������ν���Ծ���Ʒ�ֵIJⶨ�����γ�ͨ�õļ��/ɸ��ƽ̨��ؽ��˼�������⡣
������Ԫ�����ʷ������棬���ķ��[164]�����˵����ϵ��������ף�ICP-MS����ҩ������е�Ӧ�á�����������-ICP-MS�����ɶ�ø[165]�����ᰲ�淨������Ƭ[166]�еĶ����к�Ԫ��ͬʱ���вⶨ��ICP-MS������ͬʱ�ⶨ������ҩ�����еĶ���Ǩ�����ӣ������ڿ�չ�����Լ��Բ���ҩ���ĵ��ʿء���ע���������������ڵ���貣������ע���ƿ�У���ǿ�������顢��������ͳ���������ֲ�Ʒ�еĹ衢�����������ƣ�������Ҳ��һ���̶ȵ�������þ���ӡ�Ǧ���顢��Ǩ����ֵ���в������������Ե���PDEֵҪ��[167]������4%�Ĵ�����Һ�Բ���ҩ���Ľ��н��ᣬ�ⶨ����Һ�е�11��Ԫ�����ʣ��������Ľ������ϸߣ������������Ӧ�ù㷺�������������Ľ�������Ϊ����ҩ���ĵļ��ָ��[168]��
�����ڶԸ���ҩƷ��װ�����н�����/Ǩ����ķ������棬ϵͳ���о��н��ټ������ɵ�[169]����GC-MS����±�����������еĻӷ��ԳɷֺͿ���ȡ����Ϣ����Ϊ�ӷ��Գɷ��еĹѾ���Ϳ��������Է�ӳ��������������Դ���������������������ֲ�ͬ�����Ĺ軯���ղ��죬�����ӷ��Գɷ��籥���������뽺�������䷽�йأ�����ȡ��ɷ��е�S8�������ڲ�������ϵ��ʶ�������϶���о������ڶԸ����������Ӽ����ر��ǿ�����Ǩ�����ļ���ϡ������GC-MS/MS�ⶨһ����ʹ����Һ����Ʒ�����ܼ�ƫ��������������TOTM�����ܳ���[170]������LC-MS/MS�ⶨ��Ĥ������ȫ����������(PFOS)��ȫ������(PFOA)��Ǩ��[171]������GC�ⶨ������������������Ǩ��[172]�������㹲����Һ�ô�[173]��������Һ��װ����������[174]��ҩ�ö�������[175]�еĸ�������ȵIJⶨ��������GC-MS���������ù���۸��з��ֶ��ֶ����[176]�Ͷ����ڱ���������[177]����ʾ�Է�ʿ�ָ����еĸ���Ǩ����Ŀ������ڱ��С�
����3.4�� ���ʵĶ�������
�����Ի�ѧҩƷ�ж������ʵ�ʶ����ж�һֱ�����ʰ�ȫ�����ۺ��ȿ��Ƶ��ѵ㡣���ݻ�ѧҩƷ���ʶ��Ե����ʣ�Ŀǰ�ɽ����Ϊ3�ࣺMIs����ͨ�������ʺ���ͨ���ʡ���MIs��ʹ�����ʴ�Ʒ����������ϸ����ͻ�����飨Ames���飩�����������������ķ�����������ʵ��Ӧ���е����ѣ�FDA��ŷ��ҩƷ�����֣�EMA����������ڶ����ʽṹ���м����Ļ����ϣ������ö����ṹ-���Թ�ϵ��QSAR��ģ�ͣ�����ͨ�������Ԥ�������Ƿ����“��ʾ�ṹ”����������Ԥ������ʲ�����“��ʾ�ṹ”������жϸ����ʲ����л����ԣ�����жϸ����ʾ���“��ʾ�ṹ”�����ͨ��Amesʵ���������ȷ�ϣ���Amesʵ����Ϊ���ԣ������ʰ�MIs���ƣ���Ϊ���ԣ����ж��䲻���л�����[178]��
�����Է��Ŵ��������ʣ�ͨ��ϣ�������Ƿ�����ض��Ķ������ý��н�һ�����������Ա�������������ȷ���Ƿ�����Ϊ�ض��Ķ������ʽ��п��ơ����ü������in silico��Ԥ��������API��ADMET���ԵIJ��죬�����ʵĶ������ý���������һ�ֿ�����Ч�ķ����������˾��(roflumilast)[179]��������ͷ���氷(cefotiam hydrochloride)[180]������ʵ��������������й�ʳƷҩƷ�춨�о�Ժ���й�ҽѧ��ѧԺҽҩ���\���о��������������˰�����ҩ�����ʶ�������ƽ̨������LC-MS/MS�����ⶨͷ�߾����ڰ��������ڵ��������[181]������һ��ͨ��QSAR�����������˰���������������ҩ��ṹ֮��Ķ�����ϵģ�ͣ�Ԥ������ͷ�߾��ؼ������ڰ��������ڵ��������[182]��������̥���������е��»��ʡ������ʱ������ʵļ��Զ��ԣ����ö��Ա��͵IJ���˵�����ʶ��Է�Ӧ�IJ���[183]�����������˶���Ϊ���ζ����롢�ζ��ٶȡ��Թ�̼��ķ�Ӧ���ĸı䣬�������ʶԻ�����ϵͳ������[183,184]���������������ʵĸı䣬��϶���������εĹ۲�[183,185]��������ĵ�ͼ�ĸı�������ʶ�����ܵ�Ӱ�죻�ڴ˻������ܽ��ͷ�߾��ؽṹ�뼱�Զ��ԡ����ԡ�����ԵȵĹ�ϵ���ɹ��ضԹ��Ҵ���ҩ��ͷ������������ʵĶ��Խ�����Ԥ��[186]���γ���ϵͳ�ض�ͷ�߾������ʶ������۵IJ����뷽��[187]�����⣬����˶��ŵͪ��ҩ��ṹ�����Եȹ�ϵ��̽��[188]���ڴ˻����ϣ��ֽ�ϵͳ����ѧ�о��IJ��Ժͷ���Ӧ�õ������ʵĶ��������У�ͨ��������Ϣѧ������ɸѡҩ�������ʵIJ���������DEGs���Ͳ��칲�������CDEGs��������ҩ��-����ҩ��-�ź�ͨ·���ӵ������磬̽���ض��İ����ٶ��Լ����û��ƣ���������-������������磬Ѱ��ҩ�ﶾ�����õĿ��ܰз��ӣ�����ͬԴ��ģ�ͷ��ӶԽӼ�����Ԥ���ģ��������з���֮�������á�����˶�C-7λ���а���뿽ṹ��ͷ�߾��ؽṹ������[184]�������[185]�Ĺ�ϵ̽�֣�C-3λ�����ϼ��ĵ���ṹ��ͷ�߾��ؽṹ����̥����[189]�ȵĹ�ϵ̽�֣�����ת¼�鼼�������������ͷ����ͪ���ʵİ����ٽ���������[190]�����÷��ӶԽӼ�����������ͷ���氷���칹������������ͬ���칹����HAS1�е�������ý������ۣ�����Ԥ�����ǵĶ���ЧӦ[191]��Ϊ��������������ҩ�����ʵĶ������ã������ƶ��������������ṩ�˽��������
����4�� ��Ҫ��һ����ע������
������ҩƷע��ĽǶȣ���ע���ʶ�ҩ�ﰲȫ�Ե�Ӱ����Ȼ�ǵ�ǰ���ص�[192]�����ڶ�MIs�Ĺ�ע���б�Ҫ����Ʒ��ԭ��ҩ�����µķ���Ҫ�����¿�չ�ȶ����о��������еĽ��������ر��ǻӷ��Խ������ʿ�չ��������[193]����Ե�ǰ���ڽ��е�ҩƷһ�������۹�������һ����ָ�������塣Ȼ��������ÿһ��API�ĺϳ�ƽ����Ҫ6����Ӧ��ÿһ���ϳ�·��ƽ����Ҫ�õ�4�ַ�Ӧ�м��壻�ںϳɵ�����裬���±������ȣ�acid chlorides�������㰷�����˶����壨Michael acceptors���dz��õ�4�ַ�Ӧ�м��壬��API�п��ܴ��ڵ�MIs��������������Ŀ��ƣ�����һζ��Ҫ���䲻����Ӧ�������˵��ʿز���[194]���ڶ�ҩ�オ�������л����Է�������ʱ��Ҳ��Ӧ�����ݼ������/���������Ԥ��Ľ����������δ�õ�ʵ����֤�ļ��轵�������е��飬��Ӧ���ص��ע��Щ��ǿ�ƽ������顢���ٻ����ȶ����о��й۲쵽�Ľ���;������Ҫ�������[195]��Ȼ����Ŀǰ������жϡ�Ԥ��ʹ����ϳɹ������γɵ�MIs����Ȼ�Ѿ�������������������������ҩ���е�N-������[196]�������������ʴ�������ԭ��Ӧ������Ӧ�Ŀ���[197]���Թ��ɽ������ʵĴ���[198]�ȣ���Ȼû���γɽ�ͳһ�IJ��Ժͷ��������ѳ�Ϊ���ƹ��ڶ�MIs���Ƶ�ƿ����
��������ҩ�����ʵĶ��Է�Ӧ����Ϊ����Ż����������Ӧͨ·(pathway)����ȷ�������ϵ��ʹ����������ģ���ڷ���ˮƽ��̽�ֻ�����Ķ��Է�Ӧ���ƣ���������������ڻ����еIJ�����Ӧ��Ϊ���ܣ���������Ϊ��ܻ������з����������µ�ʵ�ù���[199,200]����Ҳ�����������ν�ҩ��/���ʶ���ѧ��Ϣ��ҩ���ٴ�������Ӧ���������ν�������Ӧ��Ϣ���Ʒ�����������һֱ�����������Ƶ����⡣
���������
����[1] Hu CQ. Current situation and the trend in impurity control of chemical drugs [J]. Sci China Chem (�й���ѧ����ѧ), 2010, 40: 679-687.
����[2] Hu CQ. Current situation and the trend in impurity profiling of chemical drugs [J]. Chin J New Drugs (�й���ҩ��־), 2015, 24: 1727-1734.
����[3] Olsen BA, Sreedhara A, Baertschi SW. Impurity investigations by phases of drug and product development [J]. Trends Anal Chem, 2018, 101: 17-23.
����[4] Harvey J, Fleetwood A, Ogilvie R, et al. Management of organic impurities in small molecule medicinal products: deriving safe limits for use in early development [J]. Reg Toxicol Pharmacol, 2017, 84: 116-123.
����[5] Muller L, Mauthe RJ, Riley CM, et al. A rationale for determining, testing, and controlling specific impurities in pharmaceuticals that possess potential for genotoxicity [J]. Regul Toxicol Pharmacol, 2006, 44: 198-211.
����[6] Teasdale A, Elder DP. Analytical control strategies for mutagenic impurities: current challenges and future opportunities? [J]. Trends Anal Chem, 2018, 101: 66-84.
����[7] Hussain S, Gosar A, Tabrez Shaikh T. Impurity profiling in pharmaceuticals: a review [J]. World J Pharm Res, 2018, 7: 305-320.
����[8] Snodin D, Teasdale A. Mutagenic alkyl-sulfonate impurities in sulfonic acid salts: reviewing the evidence and challenging regulatory perceptions [J]. Org Process Res Dev, 2015, 19: 1465-1485.
����[9] Gong HH, Ihle N, Jones MT, et al. Control strategy for small molecule impurities in antibody-drug conjugates[J]. AAPS PharmSciTech, 2018, 19: 971-977.
����[10] Gottlieb S. Advancing Toward the Goal of Global Approval for Generic Drugs: FDA Proposes Critical First Steps to Harmonize the Global Scientific and Technical Standards for Generic Drugs [EB/OL]. 2018 [2019-03-19]. https://www.fda.gov/NewsEvents/Newsroom/FDAVoices/ucm623665.htm.
����[11] International Conference on Harmonisation (ICH). ICH Reflection Paper: Further Opportunities for Harmonization of Standards for Generic Drugs [EB/OL]. 2019 [2019-04-19]. https://www.ich.org/products/reflection-papers.html.
����[12] G?r?g S. Critical review of reports on impurity and degradation product profiling in the last decade [J]. Trends Anal Chem, 2018, 101: 2-16.
����[13] Reddy AVB, Jaafar J, Umar K, et al. Identification, control strategies, and analytical approaches for the determination of potential genotoxic impurities in pharmaceuticals: a comprehensive review [J]. J Sep Sci, 2015, 38: 764-779.
����[14] Al Azzam KM, Aboul-Enein HY. Recent advances in analysis of hazardous genotoxic impurities in pharmaceuticals by HPLC, GC and CE [J]. J Liq Chromatographr Rel Tech, 2016, 39: 1-7.
����[15] Sun M, Liu DQ, Kord AS. A systematic method development strategy for determination of pharmaceutical genotoxic impurities [J]. Org Process Res Dev, 2010, 14: 977-985.
����[16] Liu DQ, Korda AS. Analytical challenges in stability testing for genotoxic impurities[J]. Trends Anal Chem, 2013, 49: 108-117.
����[17] McCalley DJ. Understanding and manipulating the separation in hydrophilic interaction liquid chromatography-a review [J]. J Chromatographr A, 2017, 1532: 49-71.
����[18] Jain M, Srivastava V, Kumar R, et al. Determination of five potential genotoxic impurities in dalfampridine using liquid chromatography [J]. J Pharm Biomed Anal, 2017, 133: 27-31.
����[19] Cohen RD, Liu Y, Gong X. Analysis of volatile bases by high performance liquid chromatography with aerosol-based detection [J]. J Chromatogr A, 2012, 1229: 172-179.
����[20] Denton JR, Berwick L, Loughlin TP. Development of a low level detection method for 2-chloromalonaldehyde in active pharmaceutical ingredients by HILIC separation [J]. Anal Methods, 2016, 8: 4659-4663
����[21] Dou?a M, Klva��a R, Doubsk? J, et al. HILIC-MS determination of genotoxic impurity of 2-chloro-N-(2-chloroethyl)ethanamine in the vortioxetine manufacturing process [J]. J Chromatogr Sci, 2016, 54: 119-124.
����[22] Grinberg N, Albu F, Fandrick K, et al. Assay at low ppm level of dimethyl sulfate in starting materials for API synthesis using derivatization in ionic liquid media and LC-MS [J]. J Pharm Biomed Anal, 2013, 75: 1-6.
����[23] Van Wijk AM, Niederl?nder HAG, Siebum AHG, et al. A new derivatization reagent for LC-MS/MS screening of potential genotoxic alkylation compounds [J]. J Pharm Biomed Anal, 2013, 74: 133-140.
����[24] Frenzel W, Markeviciute I. Membrane-based sample preparation for ion chromatography-Techniques, instrumental configurations and applications [J]. J Chromatographr A, 2017, 1479: 1-19.
����[25] Zhang XW, Zheng HG, Chen LL. Determination of trace hydroxylamine in rasagiline mesylate by online SPE-ion chromatography [J]. Chin Pharm J (�й�ҩѧ��־), 2017, 52: 1801-1804.
����[26] Lesellier E, West C. The many faces of packed column supercritical fluid chromatography-a critical review [J]. J Chromatographr A, 2015, 1382:2-46.
����[27] Vera CM, Shocka D, Dennis GR, et al. Contrasting selectivity between HPLC and SFC using phenyl-type stationary phases: A study on linear polynuclear aromatic hydrocarbons [J]. Microchem J, 2014, 119: 40-43.
����[28] Lecoeur M, Decaudin B, Guillotin Y, et al. Comparison of high-performance liquid chromatography and supercritical fluid chromatography using evaporative light scattering detection for the determination of plasticizers in medical devices [J]. J Chromatographr A, 2015, 1417: 104-115.
����[29] Patel I, Venkatramani CJ, Stumpf A, et al. Trace analysis of potentially mutagenic boronic acids and esters in drug substances by ICP-MS [J]. Org Proc Res Dev, 2017, 21: 182-186.
����[30] Liu DQ, Sun M, Korda AS. Recent advances in trace analysis of pharmaceutical genotoxic impurities [J]. J Pharm Biomed Anal, 2010, 51: 999-1014.
����[31] Harigaya K, Yamada H, Yaku K, et al. Novel sensitive determination method for a genotoxic alkylating agent, 4-chloro-1-butanol, in active pharmaceutical ingredients by LC-ICP-MS employing iodo derivatization [J]. Anal Sci, 2014, 30: 377-382.
����[32] Harigaya K, Yamada H, Horimoto S, et al. Sensitive quantitation of residual phenylhydrazine in antipyrine by LC-ICP-MS with iodo derivatization [J]. Anal Sci, 2014, 30: 845-850.
����[33] Pohl P, Bielawska-Pohl A, Dzimitrowicz A, et al. Impact and practicability of recently introduced requirements on elemental impurities [J]. Trends Anal Chem, 2018, 101: 43-55.
����[34] Jenke DR, Stults CL, Paskiet DM, et al. Materials in manufacturing and packaging systems as sources of elemental impurities in packaged drug products: a literature review [J]. PDA J Pharm Sci Technol, 2015, 69: 1-48.
����[35] Boetzel R, Ceszlak A, Day C, et al. An elemental impurities excipient database: a viable tool for ICH Q3D drug product risk assessment [J]. J Pharm Sci, 2018, 107: 2335-2340.
����[36] Paskiet D, Kraft C, Tullo E, et al. Assessment of Extractable Elements from Elastomers [J]. PDA J Pharm Sci Technol, 2019, 73: 83-91.
����[37] Wollein U, Bauer B, Habernegg R, et al. Potential metal impurities in active pharmaceutical substances and finished medicinal products - a market surveillance study [J]. Eur J Pharm Sci, 2015, 77: 100-105.
����[38] Li G, Schoneker D, Ulman KL, et al. Elemental impurities in pharmaceutical excipients [J]. J Pharm Sci, 2015, 104: 4197-4206.
����[39] Menoutis J, Parisi A, Verma N. Study of the use of axial viewed inductively coupled plasma atomic emission spectrometry with ultrasonic nebulization for the determination of select elemental impurities in oral drug products [J]. J Pharm Biomed Anal, 2018, 152: 12-16.
����[40] Balaram V. Recent advances in the determination of elemental impurities in pharmaceuticals – Status, challenges and moving frontiers [J]. Trends Anal Chem, 2016, 80: 83-95.
����[41] Jin C. Clean chemistry for elemental impurities analysis of pharmaceuticals in compliance with USP 232 [J]. AAPS PharmSciTech, 2015, 17: 1141-1149.
����[42] Muller AL, Oliveira JS, Mello PA, et al. Study and determination of elemental impurities by ICP-MS in active pharmaceutical ingredients using single reaction chamber digestion in compliance with USP requirements [J]. Talanta, 2015, 136: 161-169.
����[43] Da Silva CS, Pinheiro FC, Do Amaral CDB, et al. Determination of As, Cd, Hg and Pb in continuous use drugs and excipients by plasma-based techniques in compliance with the United States Pharmacopeia requirements [J]. Spectrochim Acta B, 2017, 138: 14-17.
����[44] Jenke D. Identification, analysis and safety assessment of leachables and extractables [J]. Trends Anal Chem, 2018, 101: 56–65.
����[45] Jenke D. Application of Quality by Design (QbD) Principles to Extractables/Leachables Assessment. Establishing a Design Space for Terminally Sterilized Aqueous Drug Products Stored in a Plastic Packaging System [J]. PDA J Pharm Sci Technol, 2010, 64: 527-535.
����[46] Jenke D, Carlson T. A compilation of safety impact information for extractables associated with materials used in pharmaceutical packaging, delivery, administration, and manufacturing systems [J]. PDA J Pharm Sci Technol, 2014, 68: 407-455.
����[47] Norwood DL, Paskiet D, Ruberto M, et al. Best practices for extractables and leachables in orally inhaled and nasal drug products: an overview of the PQRI recommendations [J]. Pharm Res, 2008, 25: 727-739.
����[48] Paskiet D, Jenke D, Ball D, et al. The product quality research institute (PQRI) leachables and extractables working group initiatives for parenteral and ophthalmic drug product (PODP) [J]. PDA J Pharm Sci Technol, 2013, 67: 430-447.
����[49] Jenke D. Extractables screening of polypropylene resins used in pharmaceutical packaging for safety hazards [J]. PDA J Pharm Sci Technol, 2017, 71: 346-367.
����[50] Jenke D, Couch TR, Robinson SJ, et al. The use of TOC reconciliation as a means of establishing the degree to which chromatographic screening of plastic material extracts for organic extractables is complete [J]. PDA J Pharm Sci Technol, 2014, 68: 256-270.
����[51] Zdravkovic SA, Bruss MD, Piccoli RF, et al. A method utilizing ultra-high performance liquid chromatography with ultraviolet and mass spectrometric detection for the analysis of material extracts produced during a controlled extraction study [J]. PDA J Pharm Sci Technol, 2014, 68: 504-526.
����[52] Jenke D, Egert T, Hendricker A, et al. Simulated leaching (migration) study for a model container-closure system applicable to parenteral and ophthalmic drug products [J]. PDA J Pharm Sci Technol, 2017, 71: 68-87.
����[53] Zhang DS, Xue J, Wang C, et al. Critical quality attributes of cephalosporins for injection [J]. Chin J New Drugs (�й���ҩ��־), 2016, 25: 17-24.
����[54] Chong XM, Dong X, Yao SC, et al. Research on the relationship between cephalosporin structure, solution clarity, and rubber closure compatibility using volatile components profile of butyl rubber closures [J]. Drug Dev Ind Pharm, 2019, 45: 159-167.
����[55] Deidda R, Orlandini S, Hubert P, et al. Risk-based approach for method development in pharmaceutical quality control context: a critical review [J]. J Pharm Biomed Anal, 2018, 161: 110-121.
����[56] Dispas A, Avohou HT, Lebrun P, et al. ‘Quality by design’ approach for the analysis of impurities in pharmaceutical drug products and drug substances [J]. TrAC-Trends Anal Chem, 2018, 101: 24-33.
����[57] Peraman R, Bhadraya K, Reddy YP. Analytical quality by design: a tool for regulatory flexibility and robust analytics [J]. Int J Anal Chem, 2015, 2015:1-9.
����[58] Parr MK, Schmidt AH. Life cycle management of analytical methods [J]. J Pharm Biomed Anal, 2018, 147: 506-517.
����[59] Sahu PK, Ramisetti NR, Cecchi T, et al. An overview of experimental designs in HPLC method development and validation [J]. J Pharm Biomed Anal, 2018, 147: 590-611.
����[60] Tumpa A, Staji? A, Jan?i?-Stojanovi? B, et al. Quality by design in the development of hydrophilic interaction liquid chromatography method with gradient elution for the analysis of olanzapine [J]. J Pharm Biomed Anal, 2017, 134: 18-26.
����[61] Zhang X, Hu CQ. Application of quality by design concept to develop a dual gradient elution stability-indicating method for cloxacillin forced degradation studies using combined mixture-process variable models [J]. J Chromatogr A, 2017, 1514: 44-53.
����[62] Taheri M, Moazeni-Pourasil RS, Sheikh-Olia-Lavasani M, et al. Central composite design with the help of multivariate curve resolution in loadability optimization of RP-HPLC to scale-up a binary mixture [J]. J Sep Sci, 2016, 39: 1031-1040.
����[63] Alladio E, Pirro V, Salomone A, et al. Chemometric approach to open validation protocols: Prediction of validation parameters in multi-residue ultra-high performance liquid chromatography-tandem mass spectrometry methods [J]. Anal Chim Acta, 2015, 878: 78-86.
����[64] Amos RIJ, Haddad PR, Szucs R, et al. Molecular modelling and prediction accuracy in quantitative structure-retention relationship calculations for chromatography [J]. Trends Anal Chem, 2018, 105: 352-359.
����[65] Tyteca E, Talebi M, Amos R, et al. Towards a chromatographic similarity index to establish localized quantitative structure-retention models for retention prediction: Use of retention factor ratio [J]. J Chromatogr A, 2017, 1486: 50-58.
����[66] Taraji M, Haddad PR, Amos RIJ, et al. Error measures in quantitative structure-retention relationships studies [J]. J Chromatogr A, 2017, 1524: 298-302.
����[67] Taraji M, Haddad PR, Amos RI, et al. Prediction of retention in hydrophilic interaction liquid chromatography using solute molecular descriptors based on chemical structures [J]. J Chromatogr A, 2017, 1486: 59-67.
����[68] Wen Y, Talebi M, Amos RIJ, et al. Retention prediction in reversed phase high performance liquid chromatography using quantitative structure-retention relationships applied to the hydrophobic subtraction model [J]. J Chromatogr A, 2018, 1541: 1-11.
����[69] Zhang X, Li J, Wang Chen, et al. Identification of impurities in macrolides by liquid chromatography-mass spectrometric detection and prediction of retention times of impurities by constructing quantitative structure–retention relationship (QSRR) [J]. J Pharm Biomed Anal, 2017, 145: 262-272.
����[70] Wang C, Li J, Feng YC, et al. Construction of the quantitative structure retention relationship of cefdinir related substances [J]. Acta Pharm Sin (ҩѧѧ��), 2015, 50: 1161-1166.
����[71] Zhang X, Hu CQ. Application of column characterization systems in selecting optimal RP-HPLC columns [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 942-949.
����[72] ?uvela P, Liu JJ, Plenis A, et al. Assessment of column selection systems using partial least squares[J]. J Chromatogr A, 2015, 1420: 74-82.
����[73] Andri? F, Héberger K. How to compare separation selectivity of high-performance liquid chromatographic columns properly? [J]. J Chromatogr A, 2017, 1488:45-56.
����[74] Wang J, Guo Z, Shen A, et al. Hydrophilic-subtraction model for the characterization and comparison of hydrophilic interaction liquid chromatography columns [J]. J Chromatogr A, 2015, 1398: 29-46.
����[75] Zhang WQ, Hu QX, Zhang X, et al. The selection of suitable columns for a reversed-phase liquid chromatographic separation of beta-lactam antibiotics and related substances via chromatographic column parameters [J]. J Chromatogr A, 2014, 1323: 87-96.
����[76] Zhang X��Hu CQ��Selecting optimal columns for clarithromycin impurity analysis according to the quantitative relationship of hydrophobic subtraction model [J]. J Pharm Biomed Anal, 2017, 136: 162-169.
����[77] ?uvela P, Skoczylas M, Liu J, et al. Column characterization and selection systems in reversed-phase high-performance liquid chromatography [J]. Chem Rev, 2019, 119: 3674-3729.
����[78] Robinson MW, Hill AP, Readshaw SA, et al. Use of calculated physicochemical properties to enhance quantitative response when using charged aerosol detection [J]. Anal Chem, 2017, 89: 1772-1777.
����[79] Singh DK, Sahu A, Kumar S, et al. Critical review on establishment and availability of impurity and degradation product reference standards, challenges faced by the users, recent developments, and trends [J]. Trends Anal Chem, 2018, 101: 58-107.
����[80] Li W, Zhang WQ, Li X, et al. Development and application of reference materials containing mixed degradation products of amoxicillin and ampicillin [J]. Acta Pharm Sin (ҩѧѧ��), 2014, 49: 1310-1314.
����[81] Wen HL, Jiang MH, Yang MC, et al. Study and preparation of reference materials containing mixed impurities of epirubicin hydrochloride [J]. J Chin Antibiot (�й���������־), 2017, 42: 46-51.
����[82] Hong P, Phoebe AD, Jones MD. Study of relative response factors and mass balance in forced degradation studies with liquid chromatography/photo-diode array detector/evaporative light scattering detector/mass spectrometry system [J]. J Chromatogr A, 2017, 1512: 61-70.
����[83] Liu SY, Yao SC, Zhang H, et al. Determination of relative response factors of cefazolin impurities by quantitative NMR [J]. AAPS PharmSciTech, 2017, 18: 1895-1900.
����[84] Wang LJ, Tian Y, Zhang ZJ. Overview and discussion on the impurity investigation for dihydropyridines [J]. Chin J Pharm Anal (ҩ�������־), 2018, 38: 2045-2053.
����[85] Yuan YZ, Zhang M, Hu CQ. Advances in the components analysis of aminoglycosides [J]. Chin Pharm J (�й�ҩѧ��־), 2017, 52: 1772-1779.
����[86] Yang HT, Song HJ, Wu Y, et al. Determination of related substances in celecoxib raw material by HPLC [J]. Chin J Pharm Anal (ҩ�������־), 2019, 39: 164-170.
����[87] Yang HT, Ma YL, Song HJ, et al. Determination of related substances in cinacalcet hydrochlorid raw material by HPLC [J]. Chin J Pharm Anal (ҩ�������־), 2018, 38: 844-850.
����[88] Zhang HZ, Qin F, Liu H. Analysis of the related substances and content of effective bacitracin by HPLC combined with component preparation [J]. Chin Pharm J (�й�ҩѧ��־), 2018, 53: 2041-2046.
����[89] Yang XM, Liang YK, Yu LK, et al. Qualitative and quantitative analysis of related substances in clotrimazole cream [J]. Acta Pharm Sin (ҩѧѧ��), 2018, 53: 2093-2098.
����[90] Zhu XP, Chen AP, Dai YZ. HPLC method with correction factor for determination of related substances in compound ezetimibe and rosuvastatin calcium tablets [J]. Chin Pharm J (�й�ҩѧ��־), 2017, 52: 140-146.
����[91] Yuan XJ, Yao LY, Su L, et al. Determination of related substances of anacetrapib by RP-HPLC [J]. Chin J Pharm Anal (ҩ�������־), 2019, 39: 304-309.
����[92] Yu QY, Yao K, Liu Y, et al. HPLC determination of related substances in vonoprazan fumarate [J]. Chin J Pharm Anal (ҩ�������־), 2018, 38: 728-733.
����[93] Chang Y, Tian Y, Ma Yue, et al. Determination of furbenicillin sodium and its related substances by HPLC [J]. Acta Pharm Sin (ҩѧѧ��), 2015, 50: 1632-1636.
����[94] Zhou LH, Tang D, Zhou JH, et al. Quantitative analysis of impurities in vardenafil hydrochloride orally disintegrating tablets [J]. Chin Pharm J (�й�ҩѧ��־), 2016, 51: 1790-1794.
����[95] Sharma MK, Murugesan M. Forced degradation study an essential approach to develop stability indicating method [J]. J Chromatogr Sep Tech, 2017, 8: 349.
����[96] Sengupta P, Chatterjee B, Tekade RK. Current regulatory requirements and practical approaches for stability analysis of pharmaceutical products: A comprehensive review [J]. Int J Pharm, 2018, 543: 328-344.
����[97] Ye Q, Ding W, Rinaldi F, et al. Structural characterization of low level degradants in aztreonam injection and an innovative approach to aid HPLC method validation [J]. J Pharm Biomed Anal, 2016, 124: 358-364.
����[98] Lei YP, Jin B, Li T, et al. Studies on the degradation impurity of linezolid tablets by UFLC-MS/MS [J]. Acta Pharm Sin (ҩѧѧ��), 2017, 52: 971-976.
����[99] Liu Y, Sun X, Tian Y, et al. Impurity profile study of cefradine dihydrate [J]. Chin Pharm J (�й�ҩѧ��־), 2017, 52: 1639-1643.
����[100] Zhang CY, Li J, Gao JM, et al. The impurity profiling of paclitaxel and its injection by UPLC-MS/MS [J]. Acta Pharm Sin (ҩѧѧ��), 2016, 51: 965-971.
����[101] Xue J, Zhu KX, Hu CQ. Impurity profiles comparison of cefoxitin sodium for injection [J]. J Chin Antibiot (�й���������־), 2016, 41: 606-613, 623.
����[102] Li Q, Mei Q, Liu Y. Analysis of impurities in octreotide acetate and its injection [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 492-501.
����[103] Xin J, Li W. Analysis of related substances in erythromycin estolate tablets [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 2231-2237.
����[104] Yao SC, Zhang X, Hu CQ. Quality analysis of domestic midecamycin acetate granules [J]. J Chin Antibiotics (�й���������־), 2017, 42: 496-502.
����[105] Wei NY, Zhou Y, Yu LJ, et al. Analysis of related substances in pravastatin sodium and its preparations by UPLC-DAD-MS [J]. Chin J Pharm Anal (ҩ�������־), 2018, 38: 1539-1549.
����[106] Liu ZX, Cheng QL, He L. Analysis of related substances in valsartan capsules [J]. Chin Pharm J (�й�ҩѧ��־)��2015, 50: 1624-1629.
����[107] Huang LY, Song M, Li ZH, et al. Analysis of related substances in ketorolac tromethamine and ketorolac tromethamine injection by HPLC-DAD-MS [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 1406-1422.
����[108] Li M, Hou JF, Liu ZZ, et al. Study on detection method of related substances and analysis of impurities in cisplatin injection [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 1882-1889.
����[109] Zhou YR, Zha JM, Jin ZX, et al. Determination of related substances in lidocaine aerosol by HPLC [J]. Chin J Pharm Anal (ҩ�������־), 2018, 38: 469-476.
����[110] Zhang X, Yao SC, Hu CQ. Application of design of experiment concept to develop and optimize an HPLC method to analyze the related substances in josamycin propionate [J]. J Chin Antibiot (�й���������־), 2019, 44: 376-382.
����[111] Hou JF, Li M, Li WD, et al. Identification of related substances in etimicin sulfate by HPLC-HRMS/MS [J]. Chin Pharm J (�й�ҩѧ��־), 2018, 53: 817-825.
����[112] Wang LX, Wang C, Zhang DS, et al. Quality assessment of domestic sulbenicilin sodium for injection [J]. J Chin Antibiot (�й���������־), 2017, 42: 545-549.
����[113] Xiao H, Hong JW, Peng J, et al. Quality assessment of domestic cefoxitin sodium for injection [J]. J Chin Antibiot (�й���������־), 2017, 42: 470-475.
����[114] Chong XM, Li J, Wang Y, et al. The control of the critical quality attributes of amoxicillin and clavulanate potassium tablet [J]. Acta Pharm Sin (ҩѧѧ��), 2016, 51: 1121 ?1124.
����[115] Chong XM, Wang LX, Wang C, et al. The related substance analysis and critical quality attributes control of cefradine granules [J]. Chin J New Drugs (�й���ҩ��־), 2018, 27: 74-82.
����[116] Wang L, Wu ZZ, Wang TS, et al. Correlation study between production process and impurity profile of meclofenoxate hydrochloride for injection [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 1304-1308.
����[117] Li YL, Zhou XX, Yao Y, et al. Quality assessment of ampicillin sodium and cloxacillin sodium for injection [J]. J Chin Antibiot (�й���������־), 2019, 44: 295-299.
����[118] Huo LR, Xue XT, Zhao Q, et al. Determination of isomers in ticagrelor [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 2006-2012.
����[119] Zhang X, Li H, Zhang HL, et al. Determination of enantiomer in tofacitinib citrate bulk drug and tablets by normal-phase HPLC [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 1291-1297.
����[120] Dai HX, Yang X, Lin CM. Enantioseparation of 1,4-dihydropyridines calcium antagonistsby supercritical fluid chromatography [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 1513-1519.
����[121] Jin W, Lin H, Chen Y, et al. Chiral separation of ezetimibe and R-enantiomer by supercritical fluid chromatography [J]. Chin Pharm J (�й�ҩѧ��־), 2015, 5: 68-71.
����[122] Zhao LJ, Yu J, J HD. Impurity profile study and HPLC method development for related substances determination of leuprorelin acetate [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 1423-2429.
����[123] Ren X, Tian WJ, Yang HX, et al. Structure identification of primary degradation impurities in domestic calcitonin salmon injections by LC-MS [J]. Chin Pharm J (�й�ҩѧ��־), 2015, 50: 174-177.
����[124] Li J, Song YJ, Dai CX, et al. Quantification of free sugars in glycopeptide drugs by high performance anion exchange chromatograghy-pulsed amperometric detection [J]. Chin Pharm J (�й�ҩѧ��־), 2015, 50: 1547-1552.
����[125] Zhu WX, Yang R, Xu YW, et al. Characteristics of triacylglycerols composition of medium/long chain structured triacylglycerols by ultraperformance convergence chromatography combined with a quadrupole time-of-flight mass spectrometry [J]. Chin Pharm J (�й�ҩѧ��־), 2016, 51: 1324-1329.
����[126] Xiao J, Zhu ZL, Wang RR, et al. HPLC determination of aspartate in potassium aspartate and magnesium aspartate injection and its related substances [J]. Chin J Pharm Anal (ҩ�������־), 2018, 38: 477-484.
����[127] Shen DD, Zeng J, Wang Y, et al. Determination of related substances and content of glucosamine hydrochloride by HPLC [J]. Chin Pharm J (�й�ҩѧ��־), 2017, 52: 314-318.
����[128] Li J, Zhang PP, Chong XM, et al. Analysis of polymer impurities in co-amoxicillin and potassium clavulanate preparations [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 1430-1440.
����[129] Li J, Zhang PP, Yao SC, et al. Analysis of polymer impurities in cefradine raw materials and preparations [J]. J Chin Antibiot (�й���������־), 2019, 44: 362-369.
����[130] Narayanam M, Handa T, Sharma P, et al. Critical practical aspects in the application of liquid chromatography–mass spectrometric studies for the characterization of impurities and degradation products [J]. J Pharm Biomed Anal, 2014, 87: 191-217.
����[131] Wang C, Liang RQ, Niu ZR, et al. Fragmentation pattern analysis of 7 β-adrenergic agonists by electrospray ionization mass spectrometry [J]. Chin J Pharm Anal (ҩ�������־), 2018, 38: 1419-1426.
����[132] Wang H, Huang H, Cao J, et al. Mass spectral profile for rapid differentiating beta-lactams from their ring-opened impurities [J]. Biomed Res Int, 2015, 2015:1-13.
����[133] Qian JQ, Correra TC, Li J, et al. Differentiation of Cefaclor and its delta-3 isomer by electrospray mass spectrometry, infrared multiple photon dissociation spectroscopy and theoretical calculations [J]. J Mass Spectrom, 2015, 50: 265-269.
����[134] Maggio RM, Calvo NL, Vignaduzzo SE et al. Pharmaceutical impurities and degradation products: Uses and applications of NMR techniques [J]. J Pharm Biomed Anal, 2014, 101: 102-122.
����[135] Yang F, Zhang Y, Zhang JL, et al. Preparation and identification of the doxycycline hyclate impurity eluted right after the main component described in ChP 2015 [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 695-701.
����[136] Zhang JX, Xu MQ, Chen L, et al. Analysis of impurities in rifalazil [J]. Chin Pharm J (�й�ҩѧ��־), 2015, 50: 639-645.
����[137] Zeng XF, Liu J, Song M, et al. Identification of related substances in nicergoline by HPLC-MS [J]. Acta Pharm Sin (ҩѧѧ��), 2015, 50: 1026-1031.
����[138] Wang Y, Song XJ, Zhang YP, et al. Identification for related substances of O-alkyl compounds and N-alkyl compounds in iohexol by HPLC-MS/MS [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 482-491.
����[139] Qian JQ, Chen Y. Simultaneous determination of two toxic impurities in the intermediate of dabigatran etexilate by UHPLC-MS with positive/negative ionization switching [J]. Chin Pharm J (�й�ҩѧ��־), 2016, 51: 930-934.
����[140] Xie SJ, Wang H. Identification of impurities in rosuvastatin intermediate by high performance liquid chromatography coupled with linear ion trap mass spectrometry [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 243-251.
����[141] Wang C, Cheng FJ, Lu YT, et al. Identification of the related substances of zopiclone by LC-MS techniques [J]. Chin J Pharm Anal (ҩ�������־), 2019, 39: 156-163.
����[142] Wang WJ, Zeng SP, Jiang T, et al. Qualitative analysis of an unknown impurity in levocetirizine hydrochloride tablets by LC-MS [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 252-264.
����[143] Fang YL, Lin JS, Zhu WQ, et al. Investigation study of process impurity in losartan potassium by LC-PDA-QTOF-MS [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 2224-2230.
����[144] Zhang TT, Jin B, Li T, et al. Identification of related substances in a new drug aituomode by HPLC-MS/MS [J]. Chin Pharm J (�й�ҩѧ��־), 2017, 52: 68-71.
����[145] Zhao HY, Xia JX, Wang S, et al. Application of two-dimensional UPLC-Q TOF MS technology in the study of the impurity profile of leflunomide tablets [J]. Chin J Pharm Anal (ҩ�������־), 2018, 38: 1974-1980.
����[146] Huang CY, Chen MH, Cai M, et al. Preliminary exploration and structure identification of related substances of midazolam injection by 2D-LC-IT-TOF/MS [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 688-694.
����[147] Shen YT, Ma X, Nan N. Determination of neurotoxicity impurity MPTP in pethidine hydrochloride API and injection by UPLC-MS/MS [J]. Chin J Pharm Anal (ҩ�������־), 2016, 36: 911-917.
����[148] Luan SR, Liu JY, Zhang FF, et al. Determination of cyanide in vitamin B6 and 2-thiopheneacetic acid drugs by ion chromatography with amperometric detector [J]. Chin J Pharm Anal (ҩ�������־), 2018, 38: 490-494.
����[149] Zhou T, Zeng S. Quantitative analysis of trace amount of formaldehyde and formic acid in hydrochlorothiazide API using ethanol derivative method with headspace gas chromatography [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 862-868.
����[150] Wang XF, Wang Y, Song XS, et al. Pre-column derivation HPLC method for determination of aldehydes and research on the correlation between aldehydes and peroxide value in polysorbates [J]. Chin Pharm J (�й�ҩѧ��־), 2018, 53: 1222-1229.
����[151] Liu XD, Lu F, He W. Superficial analysis of sources and control of genotoxic impurities in pharmaceutical manufacturing [J]. Pharm Care Res (ҩѧ�������о�), 2017, 17: 235-237.
����[152] Zhang J, Zhang YJ, Nie B. Advances in control strategies and methods for genotoxic impurities in drug research & development [J]. Chin J Pharm (�й�ҽҩ��ҵ��־), 2018, 49: 1203-1220.
����[153] Liu XW, Li C, Han HY, et al. Advances on genotoxic impurities of sulfonate esters in pharmaceuticals [J]. Chin J Chromatogr (ɫ��), 2018, 36: 952-961.
����[154] Xie HY, Lin YL, Zhang RL, et al. Advances in analytical methods and pre-treatment techniques for genotoxic impurities [J]. Chin J Pharm Anal (ҩ�������־), 2018, 38: 1668-1676.
����[155] Xu J. Determination of aniline potentially genotoxic impurity in rivaroxaban by HPLC [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 105-110.
����[156] Xie WF, Gao YZ, Tang XX. Determination of genotoxic impurities of 2,4-dichlorophenol and 2,4,6-trichlorophenol in pyrogallol by HPLC [J]. Chin J Pharm Anal (ҩ�������־), 2018, 38: 2110-2115.
����[157] Song F, Song XN, Wang L, et al. Determination of six aniline-like genotoxic impurities in varenicline tartrate by LC-MS [J]. Chin J Pharm Anal (ҩ�������־), 2018, 38: 130-134.
����[158] Zhang YF, Qian JQ, Wang J. Determination of trace level of genotoxic impurity N,N-dimethylaniline in flucytosine by HPLC-MS/MS [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 265-271.
����[159] Qian JQ, Zhang YF, Wang J, et al. Determination of genotoxic impurity methyl p-toluenesulfonate in two kinds of crystal forms of clopidogrel hydrogen sulfate [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 1994-1999.
����[160] Liang JM, Fu C, Chen Y, et al. Determination of genotoxic impurities in escitalopram oxalate by LC-MS/MS [J]. Chin J Mod Appl Pharm (�й��ִ�Ӧ��ҩѧ), 2016, 33: 1436-1440.
����[161] Bai PF, Li HX, Guo WM. High sensitive determination of the genotoxic impurities in olaparib by LC-MS/MS [J]. Chin J New Drugs (�й���ҩ��־), 2016, 25: 865-868.
����[162] Wang HJ, Pan HJ, Liu C, et al. Determination of the genotoxic impurity in apixaban by LC-MS [J]. Chin J Pharm (�й�ҽҩ��ҵ��־), 2015, 46: 1004-1007.
����[163] Li HX, Bai PF, Liu N, et al. Determination of a genotoxic impurity and morpholin acetic acid in carfilzomib by LC-MS/MS [J]. Chin J Pharm (�й�ҽҩ��ҵ��־), 2016, 47: 1308-1310.
����[164] Xu WF, Jin PF, Xu S, et al. Application of ICP-MS in pharmaceutical analysis [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 2123-2132.
����[165] Zhong ZH, Chen ZZ, Wang P, et al. Determination of harmful elements in lumbrokinase by microwave digestion-inductively coupled plasma mass spectrometry [J]. Chin J Pharm Anal (ҩ�������־), 2019, 39: 477-483.
����[166] Xu WW, Sun Z, Zhou FM, et al. Determination of 7 kinds of elemental impurities in desvenlafaxine hydrochloride extended-release tablets by ICP-MS [J]. Chin J Pharm Anal (ҩ�������־), 2019, 39: 319-327.
����[167] Ying ZH, Jiang HM, Jiang XL, et al. Compatibility study of pantoprazole sodium for injection and glass container [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 2094-2100.
����[168] Liu YY, Yan Min, Huang HP, et al. Simultaneous determination of 11 metal elements released from pharmaceutical glass packing materials with ICP-AES [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 712-717.
����[169] Fu M, Yuan Y, Xia HQ, et al. Analysis of composition of volatile substances in halogenated butyl rubber stoppers by GC-MS [J]. Chin J Pharm Anal (ҩ�������־), 2018, 38: 818-826.
����[170] Zhang L, Zheng J, Xu AH, et al. Research on the determination for 1,2,4-benzenetricarboxylic acid tris(2-ethylhexyl) ester released from disposable infusion set [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 2106-2111.
����[171] Kong R, Mao K, Yuan YJ, et al. Determination of migration of PFOS and PFOA in the coated stopper of QT liposome injection by LC-MS/MS [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 1314-1319.
����[172] Guo MX, Liu YH. Study on influence of migration of cyclosiloxane compound in rubber stopper on quality of injection [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 2112-2116.
����[173] Wang YW, Chen L, Feng J, et al. Determination of additives of three-layer co-extrusion bags used for infusion and the migration of additives [J]. Chin J Pharm Anal (ҩ�������־), 2016, 36: 138-143.
����[174] Li Y, Sun HM, Zhang X. Determination of antioxidants in plastic infusion packaging materials and containers and the migration to injections [J]. Chin Pharm J (�й�ҩѧ��־), 2016, 51: 1699-1705.
����[175] Feng J, Cai XY, Liu Y. Determination of antioxidant?BHT and vulcanizing agent?extractable sulphur in medicinal butyl rubber [J]. Chin J Pharm Anal (ҩ�������־), 2017, 37: 702-706.
����[176] Xiao QQ, Liu XP, Duan HX, et al. Determination of 16 polycyclic aromatic hydrocarbons in chlortetracycline hydrochloride eye ointment by GC-MS [J]. J Chin Antibiot (�й���������־), 2019, 44: 383-388.
����[177] Chen X, Duan HX, Liu XP. Determination of 15 kinds of phthalates in chlortetracycline hydrochloride eye ointment by GC-MS [J]. J Chin Antibiot (�й���������־), 2019, 44: 389-393.
����[178] Wichard JD. In silico prediction of genotoxicity [J]. Food Chem Toxicol, 2017, 106(Pt B): 595-599.
����[179] Pinheiro MS, Viana GM, Vieira BA, et al. Identification, characterization and in silico ADMET prediction of Roflumilast degradation products [J]. J Pharm Biomed Anal, 2017, 138: 126-133.
����[180] Tian Y, Han Y, Hu CQ. Impurity profile study of cefotiam hydrochloride for injection [J]. Chin J New Drugs (�й���ҩ��־), 2019, 28: 513-522.
����[181] Liu Y, Zhang J, Hu C. Validated LC-MS/MS method for simultaneous analysis of 21 cephalosporins in zebrafish for a drug toxicity study [J]. Anal Biochem, 2018, 558: 28-34.
����[182] Liu Y, Zhang X, Zhang J, et al. Construction of a quantitative structure activity relationship (QSAR) model to predict the absorption of cephalosporins in zebrafish for toxicity study [J]. Front Pharmacol, 2019, 10: 31.
����[183] Chen B, Gao ZQ, Liu Y, et al. Embryo and developmental toxicity of cefazolin sodium impurities in zebrafish [J]. Front Pharmacol, 2017, 8: 403.
����[184] Han Y, Zheng Y, Zhang J, et al. Neurobehavioral effects of cephalosporins: assessment of locomotors activity, motor and sensory development in zebrafish [J]. Front Pharmacol, 2018, 9: 160.
����[185] Han Y, Chen B, Zhang J, et al. Cardiac safety evaluation in zebrafish and in silico ADME prediction of cephalosporins with an aminothiazoyl ring at the C-7 position [J]. Toxicol Appl Pharmacol, 2018, 347: 33-44.
����[186] Qian J, Han Y, Li J, et al. Toxic effect prediction of cefatirizine amidine sodium and its impurities by structure-toxicity relationship of cephalosporins [J]. Toxicol In Vitro, 2017, 46: 137-147.
����[187] Han Y, Zhang J, Hu C, et al. In silico ADME and toxicity prediction of ceftazidime and its impurities [J]. Front Pharmacol, 2019, 10: 434.
����[188] Xiao CQ, Han Y, Liu Y, et al. Relationship between fluoroquinolone structure and neurotoxicity revealed by zebrafish neurobehavior [J]. Chem Res Toxicol, 2018, 31: 238-250.
����[189] Han Y, Zhang J, Hu C. A systematic toxicity evaluation of cephalosporins via transcriptomics in zebrafish and in silico ADMET studies [J]. Food Chem Toxicol, 2018, 116(Pt B): 264-271.
����[190] Han Y, Qian JQ, Zhang JP, et al. Structure-toxicity relationship of cefoperazone and its impurities to developing zebrafish by transcriptome and Raman analysis [J]. Toxicol Appl Pharmacol, 2017, 327: 39-51.
����[191] Tian Y, Wang, YN, Han Y, et al. Isolation, identification and in silico toxicity predictions of two isomers from cefotiam hydrochloride [J]. J Pharm Biomed Anal, 2018, 158: 425-430.
����[192] Alsante KM, Huynh-Ba KC, Baertschi SW, et al. Recent trends in product development and regulatory issues on impurities in active pharmaceutical ingredient (API) and drug products. Part 2: Safety considerations of impurities in pharmaceutical products and surveying the impurity landscape [J]. AAPS PharmSciTech, 2014, 15: 237-251.
����[193] Jamrógiewicz M. Consequences of new approach to chemical stability tests to active pharmaceutical ingredients [J]. Front Pharmacol, 2016, 7: 17.
����[194] Elder DP, Teasdale A. Is avoidance of genotoxic intermediates/impurities tenable for complex, multistep syntheses? [J]. Org Process Res Dev, 2015, 19: 1437-1446.
����[195] Kleinman MH, Elder D, Teasdale A, et al. Strategies to address mutagenic impurities derived from degradation in drug substances and drug products[J]. Org Process Res Dev, 2015, 19: 1447-1457.
����[196] Strege MA, Osborne LM, Hetrick EM, et al. Assessing the risk of formation of potential genotoxic degradation products in a small-molecule kinase inhibitor drug substance and drug product [J]. Org Process Res Dev, 2015, 19: 1458–1464.
����[197] Angelaud R, Reynolds M, Venkatramani C, et al. Manufacturing development and genotoxic impurity control strategy of the hedgehog pathway inhibitor vismodegib [J]. Org Process Res Dev, 2016, 20: 1509-1519.
����[198] Gallagher WP, Vo A. Dithiocarbamates: reagents for the removal of transition metals from organic reaction media [J]. Org Process Res Dev, 2015, 19: 1369-1373.
����[199] Adeleye Y, Andersen M, Clewell R, et al. Implementing toxicity testing in the 21st century (TT21C): Making safety decisions using toxicity pathways, and progress in a prototype risk assessment [J]. Toxicology, 2015, 332: 102-111.
����[200] Meek ME, Lipscomb JC. Gaining acceptance for the use of in vitro toxicity assays and QIVIVE in regulatory risk assessment [J]. Toxicology, 2015, 332: 112-123.