染色质的基本结构是核小体,由约147bp DNA缠绕在H2A,H2B,H3,H4各两分子形成的组蛋白八聚体构成.研究染色质的结构对于揭示真核生物基因表达调控的机理有着重要意义.但是在体内真核生物的染色质的形成会受到各种因素的干扰,在体外将DNA和组蛋白组装成核小体结构可以排除这些因素的影响来模拟核小体的形成,而且体外将一段短的DNA片断重构到组蛋白八聚体上,其核小体定位主要取决于缠绕的DNA序列特性.此外,通过染色质体外组装系统,可以将不同修饰状态的组蛋白和特定DNA模板组装成染色质结构,进而进行体外转录实验,研究组蛋白的不同修饰状态以及组蛋白变体等对基因转录的表观遗传调控作用.因此,为了更好得研究基因表达调控的机理,进行染色质的体外组装是有必要的.目前,染色质体外组装主要有两种方式:一种是采用盐透析的方法,另一种是依赖ATP及蛋白因子(dNAP I和ACF)进行的染色体体外组装.本实验构建了含目的片段601及ES1,CS1序列的质粒,运用PCR的方法获得了目的片段,利用盐透析法进行了体外组装核小体结构并用Biotin标记法和EB染色法进行了检测.
1材料与方法
1.1材料
1.1.1试验菌株
大肠杆菌DH5α由本实验室保存,重组质粒pUCCS1,pUCES1,pUC601由本实验室构建.
1.1.2试剂和仪器
质粒小提试剂盒、胶回收试剂盒购自上海生工,Taq酶、限制性内切酶,T4DNA连接酶购自TaKaRa公司,Phosphatase-alkaline-AP,CDP-Star-CDP购自罗氏公司.
1.2方法
1.2.1质粒的构建
(1)含目的片段质粒的构建DNA片段CS1和ES1分别为:
CS1:CGGGAATTTCTCGGAGATTCTCGGAGGTTCCTGAGAGGTCTTCGAAGGCTTTCAGGGATCCTTCAGAGGCTTCCAGAGGCTCTTGAGGGACCTTTGGGAATTTCCGAAGGTCCTTCA
ES1:TGCTGGCAGCACTGGTGCCCGTGCTGCCAGCACTGCTGCCAGTGCTGCCAGCACGGGCAGCAGTGCTGGCAGCACGGGCACCAGTGCTGCCAGCACGGGCACCAGTGCTGCCAGCAC GGGCAC-CAGTGGTGCCAGTGCTGGCACCAC
DNA片段化学合成后,连接到pUC19质粒的Hind III和EcoR I两酶切位点(上海生工合成克隆).
(2)含601序列重组质粒的构建用BamH I和Pst I双酶切含有601序列的pblue-script重组质粒,分离601序列,酶切体系为:质粒DNA 10μL,Buffer K 5μL,BSA 0.5μL,Bam HI(20U/μL)2μL,Pst I(10U/μL)3μL,ddH2O 29.5μL,总体系50μL,37℃酶切3h;然后将切下的601DNA序列连接到pUC19质粒的BamH I和Pst I两位点之间,酶连体系为:
601DNA序列4μL,双酶切回收后pUC19载体2.5μL,T4ligase 1.5μL,T4buffer 1.5μL,ddH2O 5.5μL,总体系15μL,16 ℃酶连20h;取4μl酶连产物转入100μL E.coli DH5a中.挑取单克隆提取质粒进行酶切、测序鉴定.
1.2.2目的片段的获得
分别设计601序列、ES1,CS1DNA片段的PCR扩增引物,由上海生工合成Biotin标记与未标记的引物,引物的序列见表1.601DNA序列PCR扩增程序为:
95 ℃ 5 min;95 ℃ 20s,53 ℃ 20s,72 ℃ 30s,32个循环;72 ℃10min;4℃保存.ES1,CS1DNA片段PCR扩增程序为:
95 ℃ 5min;95 ℃ 20s;62 ℃ 20s,72 ℃ 30s,32个循环;72℃10min;4℃保存.用PCR大量扩增获得目的片段,取5μL PCR产物加入1.5%琼脂糖凝胶进行电泳检测,其余用胶回收试剂盒回收目的片段,40μL Elution Buffer回溶.
1.2.3组蛋白八聚体表达、纯化与复性按参考文献[8]中的方法分别表达四种组蛋白,纯化后进行复性,同时装配形成组蛋白八聚体.1.2.4体外组装核小体。将纯化的组蛋白八聚体与目的DNA片段以一定的比例加入到含有2mol/LNaCl的TE缓冲液中混匀,总体系80μL,然后加入透析管(Thermo,10 000MWCO)中,放入含2mol/LNaCl的TE透析液中透析16h,在此过程中用恒流泵(HL-ZS,上海青浦西仪器厂)将TE缓冲液匀速滴入透析液中,使其中NaCl的浓度降到0.6mol/L;将透析管转入到不含有NaCl的TE缓冲液中透析3~6h.1.2.5 Biotin标记检测。
取组装后含有100ng DNA的样品进行SDS-PAGE电泳,110V,3h;然后在25mA条件下转膜36min;将转好的膜放到紫外交联仪中交联,至焦耳数降到2焦耳;加封闭液(5% SDS,125mmol/L NaCl,17mmol/L Na2HPO4,8mmol/L NaH2PO4)将膜封闭1h;加入抗体放在水平摇床摇45min;加入清洗液(100mmol/L Tris-Cl,100mmol/L NaCl,10mmol/L MgCl2)洗5次,每次10min;加底物200μL;将X光片曝光、洗片.1.2.6琼脂糖凝胶电泳。组装后样品在1.5%的琼脂糖凝胶中电泳,电泳后EB染色,凝胶成像仪拍照观察分析.
2结果与分析
2.1重组质粒的构建
将理论设计的ES1,CS1序列及601序列片段插入pUC19质粒的多克隆位点后,由于质粒含有抗氨苄青霉素的基因,所以通过在含氨苄青霉素的固体平板上进行初筛,提取质粒后利用PCR和酶切 的方法进行二次鉴定后,最后通过测序确定了构建重组质粒的正确性.
2.2目的片段的获得
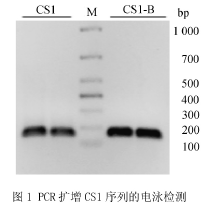
PCR扩增CS1以及标记有biotin的CS1-B目的片段,琼脂糖凝胶进行电泳检测的结果如图1所示.目的片段为157bp,从图中可以看出目的条带介于100bp~200bp之间.将所有PCR产物进行琼脂糖凝胶电泳,回收后用于组装核小体实验.2.3组蛋白八聚体的体外构建。
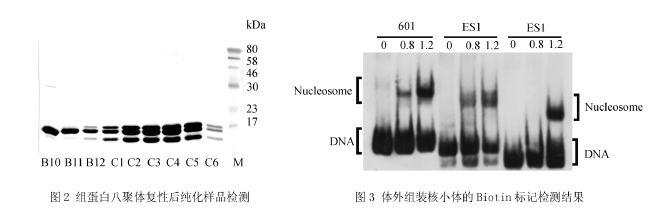
分别将4种组蛋白表达纯化后,用考马斯亮蓝法测定浓度,以等摩尔比例混合复性,用分子筛纯化,纯化后目的样品用15% SDS-PAGE电泳检测,结果如图2所示.从图中可以看出,样品C2-C5浓度较高,而且每个泳道中从上到下依次为组蛋白H3,H2A/H2B,H4,而且无杂蛋白混合,纯度较高.
2.4 Biotin标记检测核小体取组装后样品各100ng DNA进行Biotin标记检测,结果如图3所示,图中0、0.8、1.2分别为组装体系中组蛋白八聚体与DNA的质量比.从图中可看出未加组蛋白的泳道仅有DNA条带,而加入组蛋白的泳道都分别出现上下两条带,上方的条带为形成的核小体,靠下的条带为未形成核小体的游离DNA,与未加组蛋白的对照位置相同.当加入的组蛋白与DNA的比例不同时,组装核小体的效率也不同,组蛋白与DNA的比例为1.2的条带明显比0.8的条带亮.此外从图中也可以看出,序列ES1与组蛋白的结合能力差,同样比例条件下形成核小体的能力明显低于601序列与CS1序列.
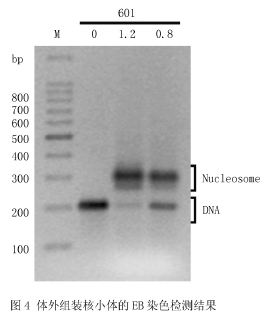
2.5 EB染色检测核小体组装后样品利用琼脂糖凝胶电泳检测结果如图4所示,图中0、0.8、1.2分别为组装体系中组蛋白八聚体与DNA的质量比.结果与利用Biotin标记检测结果类似,在加入组蛋白的样品泳道中,分别出现两条清晰的条带,上面的条带为形成的核小体,下面的条带为游离的DNA序列,而且组蛋白与DNA的组装比例越大,形成的核小体的条带越深、游离的DNA条带越浅.
3讨论
目前,染色质体外组装方式中,相对于依赖ATP及蛋白因子(dNAP I和ACF)进行的染色体体外组装,盐透析方法装配染色质系统内只含有组蛋白和DNA,不需要加入特殊的蛋白质,因此产生的染色质结构仅依赖于DNA序列的核小体定位特性.本实验以601序列为模板成功地利用盐透析方法体外组装了染色质结构,并分别运用Biotin标记及EB染色两种不同的方法进行检测.目前一般实验室EB染色能够检测到的DNA至少需要20ng以上,所以体外组装核小体结构后,直接EB染色检测电泳的灵敏度低,而且由于DNA序列缠绕到组蛋白八聚体上形成了核小体结构,也会影响EB嵌入到DNA双链中的效率,但是EB染色检测的方法也相对比较简单,无需其他过程及设备.识别Biotin的抗体专一性非常高,样品中只有微量的Biotin标记的DNA序列存在,该抗体就可以专一性的识别,所以Biotin标记检测法灵敏度较高,但检测过程较为复杂,需要进行转膜、抗体识别、底物反应、曝光等操作,费用也较EB染色昂贵.本工作为研究核小体定位、组蛋白修饰、组蛋白变体等表观遗传问题以及转录因子的结合和转录调控机制等多种生物学过程奠定了基础.致谢:组蛋白的表达、纯化与复性以及八聚体的装配在中国科学院生物物理研究所李国红课题组完成,感谢李国红研究员给予的帮助与指导.
参考文献
[1] 蔡禄,赵秀娟.核小体定位研究进展[J].生物物理学报,2009,25(6):385-395.
[2]Li G,Reinberg D.Chromatin higher-order structures and gene regulation[J].Curr Opin Genet Dev,2011,21(2):175-186.
[3]Luger K,Dechassa M L,Tremethick D J.New insights into nucleosome and chromatin structure:an ordered state or a disordered affair[J].Nat Rev Mol Cell Biol,2012,13(7):436-447.
[4]Kaplan N,Moore I K,Fondufe-Mittendorf Y,et al.The DNA-encoded nucleosome organization of a eukaryotic genome[J].Nature,2009,458(7236):362-366.
[5]Li G,Margueron R,Hu G,et al.Highly compacted chromatin formed in vitro reflects the dynamics of transcription activation in vivo[J].Molecular Cell,2010,38(1):41-53.
[6]Gracey L E,Chen Z Y,Maniar J M,et al.An in vitro-identified high-affinity nucleosome-positioning signal is capable of transiently posi-tioning a nucleosome in vivo[J].Epigenetics Chromatin,2010,3(1):13.
[7]Ito T,Bulger M,Pazin M J,et al.ACF,an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor[J].Cell,1997,90(1):145-155.
[8]Li G,Margueron R,Hu G,et al.Highly compacted chromatin formed in vitro reflects the dynamics of transcription activation in vivo[J].Molecular Cell,2010,38,41-53.