вНбЇвХДЋбЇТлЮФ

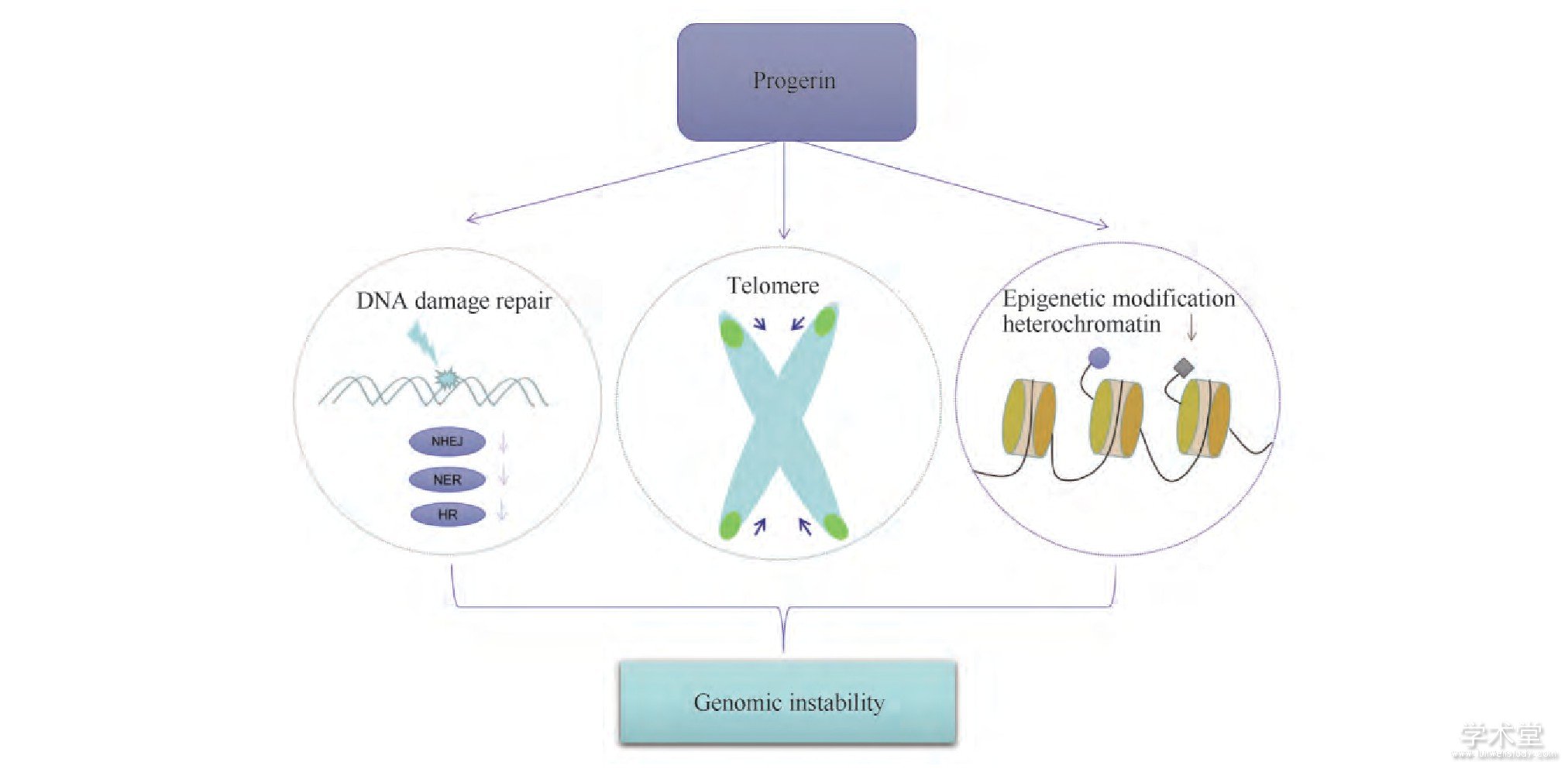
ЁЁЁЁеЊ вЊЃКЁЁЛљвђзщВЛЮШЖЈ (genomic instability) ЪЧЛњЬхЫЅРЯЕФБъжОжЎвЛ, вВЪЧЖљЭЏдчРЯжЂ (HutchinsonGilford progeria syndrome, HGPS) ЛМепЯИАћЕФЕфаЭЬиеїЁЃHGPSЕФЗЂЩњгыдчРЯЫи (progerin) ЖбЛ§УмЧаЯрЙи, ЕЋдчРЯЫиШчКЮв§Ц№ЛљвђзщВЛЮШЖЈЩаШБЗІЯЕЭГадЕФВћЪіЁЃЛљвђзщЕФНсЙЙЮШЖЈгыDNAЕФе§ШЗИДжЦЁЂDNAЫ№ЩЫаоИДЁЂЖЫСЃЕФЮЌГжКЭЮШЖЈвдМАБэЙлвХДЋбЇаоЪЮУмЧаЯрЙиЁЃБОЮФжївЊЬжТлдчРЯЫидкИФБфе§ГЃКЫЯЫВуНсЙЙЕФЛљДЁЩЯ, ЭЈЙ§гАЯьЯрЙиЭЈТЗЙиМќЕААзжЪЕФЫЎЦНЛђепЖЈЮЛ, в§Ц№ЯИАћФкбѕЛЏгІМЄдіЧПЁЂDNAИДжЦгІМЄКЭDNAЫ№ЩЫаоИДеЯА, ЯИАћDNAЫ№ЩЫдіЖрКЭЖЫСЃЕФМгЫйЫѕЖЬ, ВЂдкИФБфзщЕААзМзЛљЛЏКЭввѕЃЛЏЗНУцЕМжТЛљвђзщВЛЮШЖЈЕФЛњжЦЁЃ
ЁЁЁЁЙиМќДЪЃКЁЁдчРЯЫи; DNAЫ№ЩЫаоИД; ЛљвђзщВЛЮШЖЈ; ЖЫСЃ; БэЙлвХДЋ;
ЁЁЁЁAbstractЃКЁЁGenomic instability is one of hallmarks of aging and the typical characteristics of HutchinsonGilford progeria syndrome ( HGPS) cells. HGPS is the result of progerin accumulation. However, there is a lack of systematic explanation on how progerin causes genomic instability. The stability of genomic structure is closely related with correct DNA replication, DNA damage repair, telomere maintenance and stability, and histone epigenetic modification. This review mainly discusses the mechanism by which progerin, on the basis of structure aberration of nucleus lamina, causes intracellular oxidative stress, cellular DNA replication stress, DNA damage repair disorder, and increases cell DNA damage, telomere shortening, and dysregulation of histone methylation and acetylation, to induce genome instability by affecting the level or localization of key proteins in related pathways.
ЁЁЁЁKeywordЃКЁЁprogerin; DNA damage repair; genomic instability; telomere; epigenetics;
ЁЁЁЁЯИАћЛђепЛњЬхЕФЫЅРЯЪЧвЛИіНЅНјадЕФЙ§ГЬЃЌвВЪЧЛњЬхгаКІЮяжЪРлЛ§ЕФЙ§ГЬЁЃЫ№ЩЫЕФDNAМДЪЧгаКІЮяжЪжЎвЛЃЌРлЛ§ЕНвЛЖЈГЬЖШЛсв§Ц№ЯЕЭГЙІФмЮЩТвЃЌзюжеЕМжТЫРЭі[1]ЁЃдкГЄЦкЕФНјЛЏЙ§ГЬжаЃЌЩњЮяЬхгЕгаСЫвЛЬзГЩЪьЕФDNAЫ№ЩЫМьВтЬхЯЕЃЌвдМАеыЖдВЛЭЌЫ№ЩЫРраЭЕФDNAаоИДЗНЪНЃЌвдЮЌГжвХДЋаХЯЂЕФЭъећадКЭЮШЖЈадЁЃШчФме§ШЗаоИДЃЌЯИАћDNAНсЙЙКЭЙІФмМДФмЛжИДе§ГЃЃЌЯИАћЕУвдЮЌГже§ГЃзДЬЌ;ШєЫ№ЩЫЕФDNAВЛЭъШЋаоИДЛђепЮоЗЈаоИДЪБЃЌDNAЗЂЩњЭЛБфЃЌШОЩЋЬхЛћаЮЃЌПЩгеЕМЯИАћГіЯжЙІФмИФБфЃЌетгывЛаЉЫЅРЯЯрЙиМВВЁЕФЗЂЩњгаУмЧаСЊЯЕ[2]ЁЃЖљЭЏдчРЯжЂ (Hutchinson-Gilford progeria syndrome, HGPS) ЪЧгЩдчРЯЫи (progerin) ЕФРлЛ§Жјв§ЗЂЕФдчЫЅМВВЁЃЌВЂЧвгые§ГЃЕФЫЅРЯгааэЖрЯрЫЦжЎДІЃЌЪЧбаОПШЫРрЫЅРЯЕФРэЯыФЃаЭЁЃдкHGPSЛМепЕФГЩЯЫЮЌЯИАћжаЃЌЗЂЯжгаЯпСЃЬхЙІФмЮЩТвЁЂбѕЛЏгІМЄдіЧПЁЂДњаЛЭООЖИФБфКЭЫЅРЯЯрЙиЭЈТЗЕФМЄЛюЕШЗНУцЕФБфЛЏЃЌвдМАDNAЫЋСДЖЯСб (DNA double-strand breaks, DSBs) ЕФРлЛ§ЁЂЖЫСЃМгЫйЫѕЖЬЁЂзщЕААзБэЙлвХДЋИФБфЕШЛљвђзщВЛЮШЖЈЕФЯжЯѓЁЃБОЮФжївЊЬжТлдчРЯЫив§Ц№ЛљвђзщВЛЮШЖЈЕФЯрЙиЛњжЦ (МћFig.1) ЁЃ
ЁЁЁЁ1ЁЂ дчРЯЫиЕФаЮГЩМАКЫЯЫВуНсЙЙИФБф
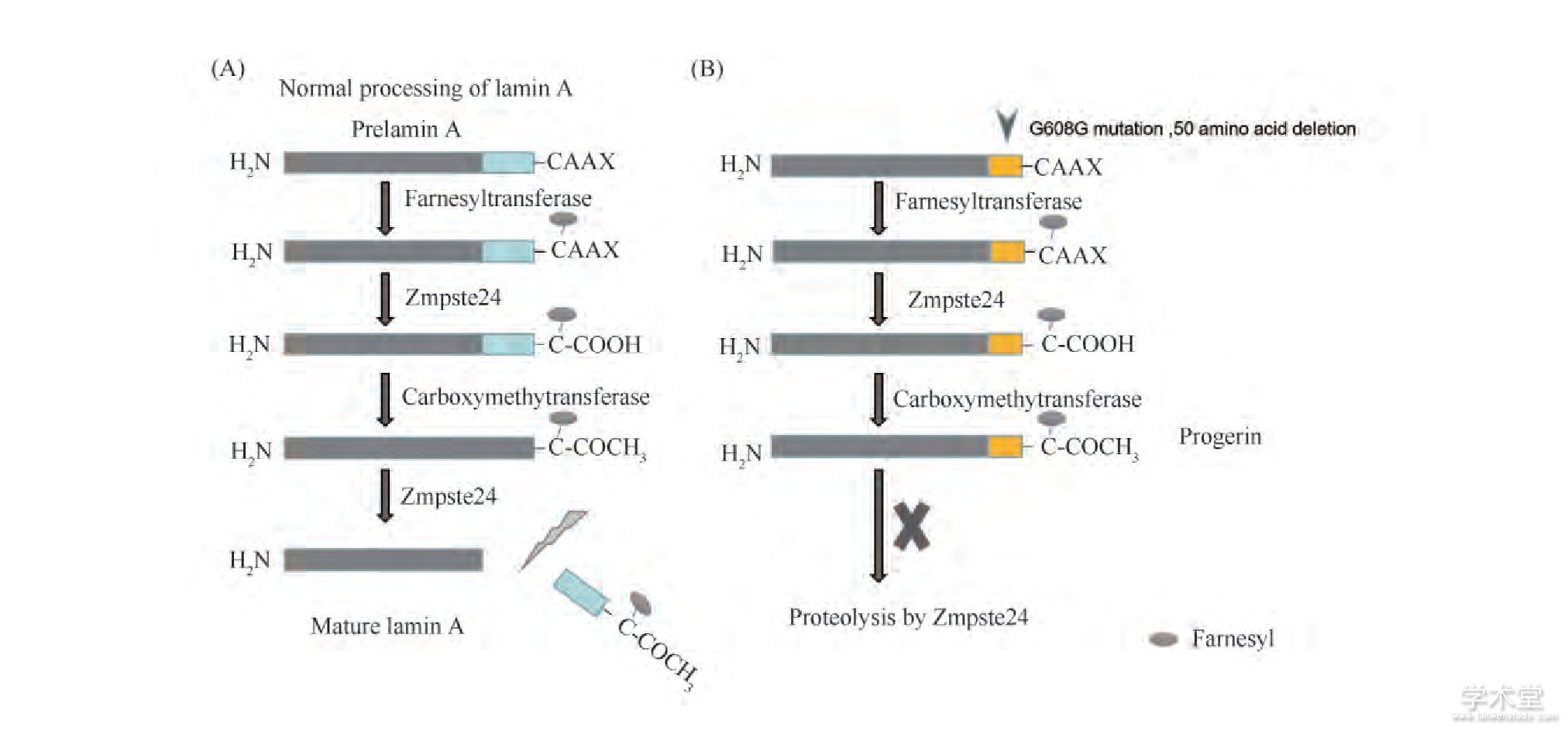
ЁЁЁЁКЫФЄЮЊФкКЫФЄКЭЯИАћжЪЬсЙЉСЫЖЏЬЌЕФБпНчЁЃКЫФЄЙІФмвдМАЯИАћКЫЕФаЮЬЌгжгыКЫЯЫВуУмЧаЯрЙиЁЃКЫЯЫВуЮЛгкФкВуКЫФЄгыШОЩЋжЪжЎМфЃЌЖдгкецКЫЯИАћЕФе§ГЃЙІФмжСЙиживЊЁЃКЫЯЫВуЕААзЪЧКЫЯЫВуЕФзщГЩГЩЗжЃЌдкКЫФкгыКЫЛљжЪЯрСЌЃЌдкКЫЭтгыжаЕШЯЫЮЌЯрСЌЃЌгаСІЕижЇГХзХЯИАћКЫКЭШОЩЋжЪЕФаЮЬЌ[3,4]ЁЃКЫЯЫВуЕААзгыКЫФЄЁЂШОЩЋжЪвдМАКЫПзИДКЯЬхдкНсЙЙКЭЙІФмЩЯгаУмЧаЙиЯЕЃЌЩцМАDNAИДжЦЁЂRNAзЊТМЁЂЯИАћКЫЕФЙЙНЈЁЂКЫПзЕФзАХфЁЂШОЩЋжЪЕФЙІФмЁЂЯИАћжмЦкКЭЕђЭіЕШЗНУц[5,6]ЁЃдкВИШщЖЏЮяжаЃЌКЫЯЫВуЕААзЗжЮЊСНжжРраЭ:AаЭКЫЯЫВуЕААз (АќРЈlamin A, lamin C, lamin C2МАlamin AΔ10) КЭBаЭКЫЯЫВуЕААз (АќРЈlamin B1, lamin B2КЭlamin B3) ЁЃAаЭКЫЯЫВуЕААзгЩLMNAЛљвђБрТыЃЌlamin B1гЩLMNB1БрТыЃЌlamin B2КЭlamin B3гЩLMNB2БрТы[7]ЁЃБрТыКЫЯЫВуЕААзЕФЛљвђЗЂЩњЭЛБфЕМжТвЛЯЕСаЕФЯИАћЙІФмИФБфЃЌЭГГЦЮЊКЫЯЫВуЕААзВЁЁЃHGPSМДЪЧвЛжжбЯжиЕФКЫЯЫВуЕААзВЁЃЌБэЯжГідчЫЅЬиеїЁЃЭЈГЃдкГіЩњКѓЕФЕквЛФъЃЌHGPSЛМепПЊЪМЯдЪОЩэВФАЋаЁЁЂЬхжиНЯЧсЁЂЭбЗЂЁЂжЌЗОЮЎЫѕЁЂгВЦЄВЁЁЂУцВПЫЅРЯКЭЙЧжЪЪшЫЩЕШвЛЯЕСаМгЫйЫЅРЯЕФЬиеїЃЌЦНОљЪйУќЮЊ13.5Ыъ[8]ЁЃбаОПБэУїЃЌHGPSЛМепГіЯждчРЯЫиЕФРлЛ§ЁЃдчРЯЫиЪЧLMNAЕФЕк11ИіЭтЯдзгЕуЭЛБфЕФВњЮяЃЌдкLMNAЛљвђЕк1 824ЮЛЕуМюЛљCЭЛБфГЩTЃЌЫфШЛетЪЧвЛИіЭЌвхЭЛБфG608GЃЌЕЋЪЧЫќМЄЛюСЫmRNAЫЎЦНЩЯЕФвЛИівўБЮМєЧаЮЛЕуЃЌдьГЩЛљвђзЊТМЕФmRNAБЛМєЧаСЫ150ИіМюЛљ (МћFig.2) ЁЃет150ИіМюЛље§КУАќКЌlamin AГЩЪьЙ§ГЬжагЩZmpste24НщЕМЕФЕААзУИЫЎНтЮЛЕуЃЌгЩгкЖЊЪЇетИіУИЧаЮЛЕуЃЌЧАЬхlamin A (prelamin A) БЛаоЪЮЕФЗЈФсЛљЭХЮоЗЈБЛЧаГ§ЖјГЩЮЊдчРЯЫиЃЌprelamin AВЛФмГЩЮЊГЩЪьЕФlamin AЁЃдчРЯЫиЕФЖбЛ§МАlamin AЕФШБЗІЃЌИФБфСЫКЫФкКЫЯЫВуЕФзщГЩКЭаЮЬЌЃЌHGPSЛМепГіЯжбЯжиЕФКЫЛћаЮЁЃКЫЯЫВуНсЙЙЩЯЕФБфЛЏв§Ц№ШОЩЋжЪЙІФмвьГЃЃЌЕМжТЛљвђБэДяИФБфЁЂЛљвђзщВЛЮШЖЈЕШвЛЯЕСаЯИАћЙІФмЕФИФБфЃЌНјЖјв§ЗЂдчЫЅ[9-11]ЁЃБОЮФжївЊЮЇШЦдчРЯЫидкИДжЦгІМЄЁЂDNAЫ№ЩЫЁЂЖЫСЃМгЫйЫѕЖЬЁЂБэЙлвХДЋИФБфЕШЛљвђзщЮШЖЈадЗНУцЕФЯрЙизїгУЛњжЦеЙПЊзлЪіЁЃ

ЁЁЁЁ2ЁЂ дчРЯЫиЕМжТИДжЦгІМЄКЭDNAЫ№ЩЫдіМг
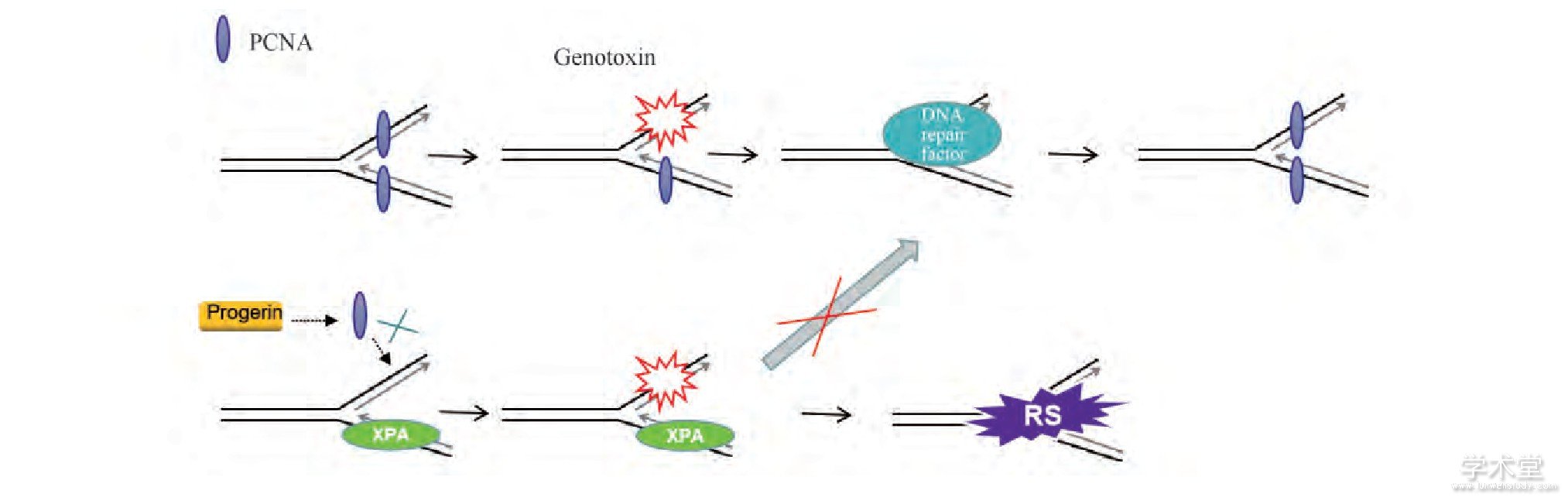
ЁЁЁЁе§ГЃЧщПіЯТЃЌlamin AПЩвдгыдіжГЯИАћКЫПЙд (proliferating cell nuclear antigen, PCNA) ЯрЛЅзїгУЃЌжиаТЦєЖЏЭЃжЭЕФИДжЦВцЃЌДгЖјЙВЭЌЮЌЛЄDNAИДжЦЕФе§ГЃНјаа[12]ЁЃPCNAЪЧDNAИДжЦЙ§ГЬжаЕФЛЌЖЏЙЬЖЈЕААзжЪЃЌдкИДжЦВцДІеаФМDNAОлКЯУИЃЌВЂЧвПЩвдМгЧПDNAГжајКЯГЩФмСІЃЌЫќЖде§ГЃЕФDNAИДжЦЁЂКЯГЩКЭбгЩьЙ§ГЬЪЧБиВЛПЩЩйЕФЁЃдкдчРЯЫиДцдкЕФЧщПіЯТЃЌPCNAЮоЗЈНсКЯдкИДжЦВцЩЯЃЌЦЦЛЕDNAИДжЦЕФНјГЬЃЌЕМжТИДжЦВцЕФЭЃжЭКЭКЫУИНщЕМЕФИДжЦВцЕФНЕНтЃЌзюжев§Ц№ИДжЦгІМЄ (replication stress, RS) [13]ЁЃдкЗжСбЕФЯИАћжаЃЌздЗЂЕФDSBsжївЊРДдДгкDNAИДжЦЃЌвђДЫЃЌдчРЯЫив§ЗЂЕФИДжЦгІМЄПЩФмЪЧHGPSЛМепЯИАћжаDNAЫ№ЩЫЕФжївЊдвђ[14]ЁЃДЫЭтЃЌHGPSЛМепЯИАћДњаЛВњЩњЕФЛљвђЖОЫи (genotoxin) ЃЌШчЯпСЃЬхбѕЛЏгІМЄЗДгІВњЩњЕФЛюадбѕздгЩЛљ (ROS) ЕШЃЌвВЛсдьГЩDNAЫ№ЩЫЕФдіЖр[2]ЁЃKubbenЕШЗЂЯжЃЌдкHGPSЛМепЕФЯИАћжаЃЌгаNRF2 (nuclear factor[erythroidderived 2]-like 2) ПЙбѕЛЏЭЈТЗЕФвжжЦ[15]ЁЃNRF2ВЮгыЕїПиNADPHЩњГЩЕФ4ИіЛљвђ (ЦЯЬбЬЧ-6-СзЫсЭбЧтУИЁЂ6-СзЫсЦЯЬбЬЧЫсЭбЧтУИЁЂЦЛЙћЫсУИ1КЭвьФћУЪЫсЭбЧтУИ1) ЕФБэДяЃЌЪЧДйНјПЙбѕЛЏЗДгІЕФЙиМќвђзг[16]ЁЃдчРЯЫиЭЈЙ§ИєРыNRF2ЪЙЦфКЫЖЈЮЛДэЮѓЃЌЕМжТNRF2зЊТМЛюадЪмЫ№ЃЌдьГЩСЫHGPSЯИАћФкГЄЦкЕФбѕЛЏгІМЄЁЃЖјдкHGPSЛМепЯИАћжаЃЌжиаТМЄЛюNRF2МѕЩйСЫDNAЫ№ЩЫБъжОЮяγ-H2AXЕФЫЎЦН[15]ЁЃ
ЁЁЁЁзїЮЊDNAЫ№ЩЫгІД№ЕФвЛВПЗжЃЌЖржжDSBsаоИДЕААзжЪПЩвдБЛеаФМЕНDSBsЫ№ЩЫВПЮЛНјаааоИДЁЃЖјдкHGPSЛМепЕФЯИАћжаЃЌдчРЯЫиЦЦЛЕDSBsДІЖдаоИДЕААзжЪЕФеаФМЃЌДгЖјЮоЗЈаоИДЪмЫ№ЕФDNAЁЃдкZmpste24-/-аЁЪѓжаЃЌвВЗЂЯжРрЫЦЕФЧщПі[17]ЁЃдкдчРЯЫиДцдкЧщПіЯТЃЌЩЋЫиГСЛ§ЦЄЗєИЩдяжЂAЛљвђ (xeroderma pigmentosum group A, XPA) ДэЮѓЖЈЮЛгкдчРЯЫив§ЗЂЕФDNAЫЋСДЖЯСбЧјгђЃЌзшЕВСЫе§ГЃЕФаоИДЕААзжЪдкDSBsВПЮЛЕФеаФМЃЌЫ№ЩЫЕФDNAЮоЗЈБЛаоИДЃЌЕМжТDNAЫЋСДЖЯСбРлЛ§діМгКЭЯИАћжадчЦкИДжЦЕФЭЃжЭ[13] (МћFig.3) ЁЃXPAЪЧКЫЫсЧаГ§аоИД (nucleotide excision repair, NER) жаЬивьЧвБиВЛПЩЩйЕФвђзгЃЌЫќПЩвддкКЫЫсЧаГ§аоИДжаЪЖБ№DNAЫ№ЩЫЃЌВЂЧвдкКЫЫсЧаГ§аоИДжаКЫУИЕФеаФМКЭКѓајЕФЮШЖЈаоИДЗЂЛгживЊЕФзїгУЃЌЕЋЪЧЫќВЂВЛЩцМАDSBsЕФаоИДЙ§ГЬ[18]ЁЃетбљвЛРДЃЌXPAгыDSBsЕФДэЮѓНсКЯЃЌЕМжТе§ГЃЕФDSBsаоИДЕААзжЪЮоЗЈБЛеаФМЕНDSBsДІЃЌЭЌЪБгжЦЦЛЕСЫКЫЫсЧаГ§аоИДЕФЭЈТЗЃЌжТЪЙдкЭЌбљЕФЭтНчЬѕМўЕФДЬМЄЯТЃЌHGPSЛМепЕФЯИАћЖдDNAЫ№ЩЫИќМгУєИаЁЃЭЌЪБЗЂЯжЃЌдкHGPSЛМепЕФЯИАћжаЃЌγ-H2AXКЭXPAЙВЭЌЖЈЮЛгкDNAИДжЦЕААзжЪЧјгђЃЌАЕЪОзХDSBsЕФВњЩњПЩФмРДздгкЭЃжЭЕФЛђепЬЎЫњЕФИДжЦВц[13]ЁЃдчРЯЫив§Ц№ИДжЦгІМЄвдМАбѕЛЏгІМЄЕФдіЧПЃЌЭЌЪБгжЦЦЛЕСЫЯИАћФке§ГЃЕФDNAЫ№ЩЫаоИДЛњжЦЃЌМгЫйСЫDNAЫ№ЩЫЕФВњЩњЁЃ
ЁЁЁЁFig.1 Genomic instability caused by progerin
ЁЁЁЁFig.2Maturation of lamin A and formation of progerin or LAΔ50
ЁЁЁЁзюНќЃЌдкВИШщЖЏЮяжаЗЂЯжСЫвЛжжКЫЫсзЊвЦУИЃЌЫќЪЧаТаЭЕФDNAИаЪмЦї (DNA sensor) МДcGAS (cGMP-AMP synthase) ЃЌФмВњЩњФкдДадЕФcGAMP (cyclic dinucleotide 2'cyc-cyclic GMP-AMP) ЃЌМЬЖјМЄЛюИЩШХЫиДЬМЄЛљвђ (stimulator of interferon genes, STING) ЁЃSTINGЪЧDNAИаЪмЭЈТЗЕФвЛИіЙиМќЕААзжЪЃЌдкУтвпЦєЖЏЗНУцгаживЊвтвхЃЌЫќвВПЩвджБНгЛђМфНгЪЖБ№dsDNA[19-22]ЁЃDNAЫ№ЩЫКЭИДжЦгІМЄВЛНіЪЧЕМжТЫЅРЯКЭАЉжЂЕФЛљвђзщВЛЮШЖЈадЕФЛљДЁЃЌЖјЧвЛЙгажњгкМЄЛюбзжЂЗДгІЁЃдкФГаЉЧщПіЯТЃЌДйНјЖёаджзСіКЭЦфЫћЫЅРЯЯрЙиМВВЁ[23]ЕФЗЂеЙЁЃР§ШчЃЌдчРЯЫиЭЈЙ§ИДжЦгІМЄв§Ц№СЫЯИАћжЪжаСуЩЂЕФКЫЫсЕФЛ§РлЃЌБЛФЃЪНЪЖБ№ЪмЬх (pattern recognition receptors, PRRs) ДэЮѓЕиЪЖБ№ЮЊЭтРДЮяЃЌМЄЛюСЫУтвпгІД№ЁЃcGAS-STNGАћжЪЕФDNAМьВтЭООЖЖдМЄЛюИЩШХЫиЗДгІжСЙиживЊЃЌВЂгыжзСіКЭЫЅРЯгаЙи[24,25]ЁЃKreienkampЕШ[26]ЖдHGPSЛМепЕФЯИАћНјааШЋЛљвђзщБэДяЗжЮіЗЂЯжЃЌЯИАћжагазЊТМаХКХзЊЕМгыМЄЛюМС1 (signal transducer and activator of transcription 1, STAT1) НщЕМЕФIFNбљгІД№ЭООЖЕФЧПСвМЄЛюЃЌвдМАГЌЙ§40ИіSTATЕїНкЕФИЩШХЫиДЬМЄЛљвђ (interferon stimulation genes, ISGs) ЕФЩЯЕїЃЌетаЉДњБэСЫЧПСвЕФИЩШХЫибљгІД№ЁЃдчРЯЫиЦЦЛЕDNAЕФИДжЦЙ§ГЬЃЌИДжЦгІМЄВњЩњЕФDSBsКЭЩЂТфдкАћжЪжаЕФКЫмеЫсЃЌМЄЛюcGAS-STINGАћжЪФкDNAИаЪмаХКХЭООЖЃЌНјЖјМЄЛюSTAT-1ЕїНкЕФИЩШХЫибљгІД№ЃЌЕМжТЯИАћФкЙЬгаУтвпЗДгІЕФЗЂЩњЃЌПЩФмВЮгыЯИАћЙІФмШБЯнКЭдчРЯЕФБэаЭЁЃ
ЁЁЁЁFig.3 A proposed model showing that DNA double-strand break repair activity is impaired in HGPS
ЁЁЁЁSTATНщЕМЕФЖдISGsЕФЕїПиЃЌМЄЛюбзжЂЕФЗЂЩњЭООЖЃЌв§Ц№ШчЖЏТіжрбљгВЛЏЕШбзжЂадгІД№ЃЌВТВтетвЛЙ§ГЬКЭдчРЯжЂЛМепвзЫРгкЙкзДЖЏТігВЛЏадаФдрВЁгаУмЧаЙиЯЕ[27]ЁЃдчРЯЫив§Ц№ИДжЦгІМЄКЭКЫУИНщЕМЕФИДжЦВцЕФНЕНтЃЌНвЪОдкHGPSЛМепЕФЯИАћжаDNAЕФЫ№ЩЫЗДгІЃЌИДжЦгІМЄАщЫцзХcGAS-STNGЭООЖЕФЩЯЕїЃЌвдМАМЄСвЕФгЩSTATЕїПиЕФИЩШХЫибљгІД№ЕФМЄЛюЃЌетПЩФмЪЧдчРЯЫив§Ц№дчЫЅЕФВЁвђбЇжЎвЛ[26]ЁЃ
ЁЁЁЁ3ЁЂ дчРЯЫив§ЗЂЖЫСЃМгЫйЫѕЖЬ
ЁЁЁЁЖЫСЃ (telomere) ЪЧДцдкгкецКЫЯИАћЯпзДШОЩЋЬхФЉЖЫЕФвЛаЁЖЮDNA-ЕААзжЪИДКЯЬхЃЌЫќгыЖЫСЃЕААзИДКЯЬх (shelterin complex) вЛЦ№ЙЙГЩСЫЬиЪтЕФ“УБзг”НсЙЙЃЌзїгУЪЧБЃГжШОЩЋЬхЕФЭъећадКЭПижЦЯИАћЗжСбжмЦкЁЃЖЫСЃЕААзИДКЯЬхАќРЈЖЫСЃжиИДађСаНсКЯвђзг1 (telomeric repeat-binding factor 1, TRF1) КЭЖЫСЃжиИДађСаНсКЯвђзг2 (telomeric repeat-binding factor 2, TRF2) ЃЌЫќУЧЕФЙІФмжЎвЛЪЧБЃЛЄШОЩЋЬхФЉЖЫВЛБЛЯИАћФкМрЖНЛњжЦЪЖБ№ЮЊDSBs[28]ЁЃдке§ГЃЯИАћжаЃЌЫцзХИДжЦДЮЪ§ЕФдіМгЃЌЖЫСЃЫѕЖЬЕНвЛЖЈГЬЖШПЩвдМЄЛюDNAЫ№ЩЫаХКХЃЌДгЖјв§ЗЂгРдЖЕФЩњГЄзшжЭЃЌМДИДжЦадЫЅРЯ[29]ЁЃЬхЭтХрбјбаОПЯдЪОЃЌдке§ГЃЕФЫЅРЯЯИАћжаЃЌдчРЯЫиЕФmRNAЫЎЦНЪЧЩ§ИпЕФ[30]ЃЌдчРЯЫиЕФРлЛ§вВЛсЕМжТЯИАћдчРЯЃЌФЧУДЖўепжЎМфДцдкЪВУДСЊЯЕ?CaoЕШ[31]ШЯЮЊЃЌдке§ГЃЕФГЩЯЫЮЌЯИАћЫЅРЯЙ§ГЬжаЃЌГжајадЕФЖЫСЃЪмЫ№ЪЧдчРЯЫиВњЩњЕФдвђжЎвЛЁЃдкЯИАћЫЅРЯЕФЙ§ГЬжаЃЌЖЫСЃЙІФмЮЩТвв§ЗЂЯИАћФкЖржжЛљвђПЩБфМєЧаЮЛЕуЕФИФБфЃЌЦфжаОЭАќРЈLMNAЛљвђЕФЭЛБфЁЃЫћУЧШЯЮЊЃЌЖЫСЃЕФж№НЅЖЊЪЇЪЧвЛжжЩЯгЮаХКХЃЌетИіаХКХМЄЛюLMNAвўБЮЕФМєЧаЮЛЕуЃЌЕМжТдчРЯЫиЕФВњЩњЁЃЗДжЎЃЌдкГЩЯЫЮЌЯИАћжаЃЌЧПжЦадбгГЄЖЫСЃЃЌвжжЦдчРЯЫиЕФВњЩњЁЃдкгРЩњЛЏЯИАћжа (гЕгаЖЫСЃУИЃЌЖЫСЃГЄЖШЮШЖЈ) ЃЌдчРЯЫиЕФmRNAБэДяЫЎЦНЪЧЯдзХНЕЕЭЕФЁЃ
ЁЁЁЁСэвЛЗНУцЃЌдкHGPSЛМепЕФЯИАћжаБэЯжгаЖЫСЃDNAЫ№ЩЫаХКХЕФдіМгЃЌЧвЯИАћжаЖЫСЃЪЧЫѕЖЬЕФ[32,33]ЁЃдке§ГЃЕФГЩЯЫЮЌЯИАћжаЃЌЭтдДадБэДядчРЯЫиЃЌЕМжТЖЫСЃВПЮЛЕФDNAЫ№ЩЫКЭвдЩњГЄзшжЭЮЊЬиеїЕФЫЅРЯЯжЯѓЁЃСэгабаОПЗЂЯжЃЌдкLMNAЭЛБфЕФГЩЯЫЮЌЯИАћжаЃЌЙЙГЩЖЫСЃИДКЯЬхЕФЖЫСЃНсКЯЕААзДѓСПМѕЩйЃЌЦфжажївЊЪЧTRF2ЕФЫЎЦН[34]ЁЃTRF2ЕФМѕЩйЦЦЛЕЖЫСЃЕФt-loopsНсЙЙЕФаЮГЩЁЃt-loopsНсЙЙЪЧБЃЛЄDNAЖЫСЃФЉЖЫУтЪмDSBаоИДЕФЙиМќвђЫиЁЃвђДЫЃЌЖдЖЫСЃЕФБЃЛЄадЯТНЕЃЌв§ЗЂЖЫСЃЫѕЖЬЃЌDNAЫ№ЩЫгІД№ЃЌШОЩЋЬхВЛЮШЖЈКЭдчЫЅ[35]ЁЃИќНјвЛВНЗЂЯжЃЌTRF2ПЩгыlamin AЛђепЦфЫћЕФКЫЯЫВуЯрЙиЕААзжЪНсКЯЃЌlamin AКЭTRF2НсКЯМгЙЬСЫt-loopsЕФаЮГЩ[36]ЃЌдчРЯЫиШДЮоДЫзїгУЁЃHGPSЛМепЕФЯИАћжаЃЌЗЂЯжгаЖЫСЃDNAЫ№ЩЫаХКХвдМАЖЫСЃМгЫйЫѕЖЬЁЃЖЫСЃЙІФмЮЩТвШчКЮЕМжТдчРЯЫиЕФВњЩњЃЌЛђепЪЧЦфжаИќгаЯъОЁЕФЯрЛЅзїгУЃЌШдШЛЪЧвЛИіУдЃЌИќЩюШыЕФЬНОПЖўепжЎМфЕФЙиЯЕНЋЖдЫЅРЯКЭЫЅРЯЯрЙиМВВЁгаИќЩюШыЕФРэНтЁЃ
ЁЁЁЁ4ЁЂ дчРЯЫигыБэЙлвХДЋИФБф
ЁЁЁЁ4.1ЁЂ дчРЯЫигАЯьзщЕААзМзЛљЛЏ
ЁЁЁЁШОЩЋжЪИФБфЪЧЫЅРЯЕФживЊдвђжЎвЛЁЃвьШОЩЋжЪЮШЖЈадЕФЮЌГжЖдЯИАћЭъећаджСЙиживЊЃЌЦфЖЊЪЇЛсЕМжТЛљвђзщВЛЮШЖЈКЭЯИАћРЯЛЏ[37]ЁЃШОЩЋжЪНсЙЙИФБфгЩDNAМзЛљЛЏКЭзщЕААзЗвыКѓаоЪЮЕШЙВЭЌНщЕМЃЌетаЉБэЙлвХДЋЕїПидкЛљвђзщЕФЮШЖЈадЁЂЛљвђБэДяЁЂЯИАћНЁПЕгыМВВЁЕШЗНУцгаУмЧаЙиЯЕ[38,39]ЁЃдке§ГЃЕФЫЅРЯЯИАћбаОПЗЂЯжЃЌH3K9me3КЭH3K27me3ЕФЫЎЦНж№НЅЯТНЕЃЌетСНжжзщЕААзЖМгаДйНјвьШОЩЋжЪФ§МЏзїгУЁЃНјвЛВНбаОПЗЂЯжЃЌЫцзХЯИАћЫЅРЯЃЌHP1α (heterochromatin protein 1 homologue-α) ЁЂNuRDИДКЯЮя (nucleosome remodelling and deacetylase chromatin remodelling complex) КЭЖрЪсЕААзМвзх (polycombgroup proteins, PCG) ЕФВПЗжЕААзжЪЃЌШчEZH2ЯТЕїЁЃHP1αЪЧвЛжжвьШОЩЋжЪНсКЯЕААзжЪЃЌЫќгыH3K9me3НсКЯЪЧНсЙЙвьШОЩЋжЪЕФЬиеїжЎвЛЃЌдкЮЌГжвьШОЩЋжЪЙІФмЗНУцЗЂЛгЙиМќзїгУ[40]ЁЃNuRDИДКЯЮяКЭH3K27МзЛљзЊвЦУИEZH2ЕФБэДяМѕЩйЃЌдьГЩH3K27me3ЕФНЕЕЭЃЌетаЉЕААзжЪЕФЯТЕїКЭвьШОЩЋжЪЖЊЪЇгаУмЧаЙиЯЕЃЌЧвЖМЪЧБэЙлГСФЌзг[41-43]ЁЃетаЉБэЙлвХДЋИФБфКЭЫЅРЯЯЂЯЂЯрЙиЃЌгабаОПЗЂЯжЃЌдкЯпГцжаЧУГ§H3K27ШЅМзЛљЛЏУИUTX1ЃЌдіМгСЫЯпГцдРДЪйУќЕФ30%[44]ЁЃ
ЁЁЁЁдкHGPSЛМепжавВБЈЕРгавдЩЯЯжЯѓЁЃбаОПЗЂЯжЃЌдчРЯЫигАЯьСЫЖрЪсЕААзМвзхВПЗжЕААзжЪЕФЖЈЮЛЃЌдьГЩH3K9me3дкЛљвђзщЩЯЕФЙуЗКЖЊЪЇЃЌЖјзщЕААзаоЪЮвьГЃЛђепМѕЩйЛсЕМжТDNAЫ№ЩЫаоИД (DNA damage repair, DDR) ШБЯн[45]ЁЃР§ШчЃЌдкDNAЫ№ЩЫаоИДЭЈТЗжаЃЌATMЪЧγ-H2AXЕФживЊЩЯгЮМЄУИЃЌγ-H2AXЭЈЙ§еаФМЯТгЮЕФаоИДЕААзжЪДгЖјНјааМАЪБЕФDNAЫ№ЩЫаоИДЁЃZhangЕШ[46]ЗЂЯжЃЌдке§ГЃЯИАћжаЃЌгУАЂУЙЫигеЕМDNAЫ№ЩЫЃЌγ-H2AXКЭH3K9me3ЫЎЦНГЩе§ЯрЙиЁЃетЪЧвђЮЊH3K9me3ЖдATMЕФМЄЛюжСЙиживЊЃЌЭЈЙ§МѕЩйH3K9ЕФМзЛљзЊвЦУИШчSUV39h1НЕЕЭH3K9me3ЫЎЦНЃЌПЩвдзшжЙDSBsЭЈТЗжаATMЕФМЄЛю[47,48]ЁЃHP1αЪЧвьШОЩЋжЪH3K9ЕФжївЊХфЬх[49]ЁЃдке§ГЃЯИАћжаЃЌH3K9КЭHP1αИЛМЏдкКЫЯЫВуЕААзЩЯЃЌВЮгыDNAЫ№ЩЫаоИДЛљвђЕФБэДяМЄЛюЃЌЙВЭЌЮЌГжЛљвђзщЕФЮШЖЈ[50]ЁЃгІД№DNAЫ№ЩЫЪБЃЌHP1αДйНјSUV39h1ЕФеаФМЃЌДгЖјдіМгH3K9me3ЫЎЦНЃЌВЂЧвЦ№ЪМDDRаХКХЁЃHGPSЛМепЕФЯИАћжаЃЌдчРЯЫиЮоЗЈЗЂЛгКЫЯЫВуЕААзЕФе§ГЃЙІФмЃЌЕМжТHP1αЙІФмЪмЫ№ЃЌгАЯьSUV39h1ЕФеаФМЃЌЦЦЛЕDNAаоИДЙ§ГЬжааоИДЕААзжЪЕФеаФМЃЌЕМжТЛљвђзщЮШЖЈадЯТНЕ[46]ЁЃгыДЫЯрЗДЃЌгаШЫШЯЮЊдкHGPSЛМепжаЃЌH3K9me3ЕФЫЎЦНЩ§ИпзшжЙСЫDDR[51]ЁЃDNAЫ№ЩЫЪБжТУмЕФШОЩЋжЪЬиБ№ЪЧвьШОЩЋжЪЃЌашвЊОгЩATMНщЕМЕФKRAB-ЯрЙиЕААз1 (KRAB-associated protein-1, KAP1ЃЌвВГЦЮЊTrim28ЛђTif1b) ЕФСзЫсЛЏЃЌНјааЙуЗКЕФКЫаЁЬхЕФжиЫмЃЌДгЖјЪшЫЩвьШОЩЋжЪЕФжТУмНсЙЙЃЌШУDNAаоИДЕААзжЪИќвзНсКЯдкЫ№ЩЫВПЮЛНјаааоИД[52]ЁЃдке§ГЃЯИАћжаЃЌlamin AКЭаэЖрЯИАћКЫЕААзжЪЯрЛЅзїгУЃЌР§ШчRbЁЂ53BP1ЁЂING1ЕШЃЌзшжЙЫќУЧБЛЕААзУИНЕНтЁЃLamin AвВПЩвдКЭSUV39h1ЯрЛЅзїгУЃЌЗРжЙЦфНЕНтЃЌдіЧПЦфЮШЖЈадЁЃЖјдчРЯЫиКЭSUV39h1ЯрЛЅзїгУЕФЧзКЭСІЧПгкlamin AЃЌЪЙЦфИќВЛвзНЕНтЁЃгЩгкдчРЯЫиНЕЕЭSUV39h1ЕФНЕНтЃЌЪЙЕУЯИАћжаH3K9me3ЫЎЦНЩ§ИпЃЌдьГЩH3K9me3НщЕМЕФвьШОжЪФ§МЏГЬЖШдіМгЃЌШОЩЋжЪНєУмЃЌвђДЫВЛвзеаФМаоИДЕААзжЪЃЌDNAаоИДеЯАЃЌНјЖјЛљвђзщЮШЖЈадЯТНЕЃЌМгЫйдчЫЅЁЃНЕЕЭSUV39h1ЫЎЦНЃЌеќОШСЫDNAаоИДКЭдчЦкЫЅРЯЃЌбгГЄZmpste24-/-аЁЪѓЕФЪйУќЁЃвдЩЯНсТлВЛЭЌЕФдвђПЩФмЪЧЪЕбщжагІгУЕФЯИАћДЋДњЪ§ВЛЭЌЃЌвдМАЯИАћДІгкВЛЭЌЕФЯИАћжмЦкЫљжТЃЌЫљвдбаОПH3K9me3КЭSUV39h1гыЫЅРЯжЎМфЕФЙиЯЕЃЌБиаыНЋЯИАћДЋДњДЮЪ§зХжиПМТЧНјШЅЁЃдчРЯЫиЭЈЙ§гАЯьHP1αЁЂEZH2ЁЂSUV39h1ЕШЕААзжЪИФБфH3K9ЕФБэЙлвХДЋаоЪЮЃЌЭЈЙ§гАЯьШОЩЋжЪНсЙЙКЭDDRЕМжТЛљвђзщЮШЖЈадЯТНЕЃЌв§Ц№дчЫЅЁЃ
ЁЁЁЁ4.2ЁЂ дчРЯЫигАЯьзщЕААзввѕЃЛЏ
ЁЁЁЁдкHGPSЛМепЕФЯИАћжаЃЌЙуЗКЕФзщЕААзМзЛљЛЏЖЊЪЇвВАщЫцзХH2BКЭH4ЕФЕЭввѕЃЛЏЃЌетПЩФмЪЧгЩгкзщЕААзввѕЃзЊвЦУИKAT8дкКЫЯЫВуЩЯЕФМѕЩйв§ЫљжТ[2]ЁЃH4K16ЕФввѕЃЛЏПЩвдДйНјЗЧЭЌдДадФЉЖЫНгКЯ (non-homologous end joining, NHEJ) КЭЭЌдДжизщаоИД (homologous recombination repair, HR) ЭООЖЃЌввѕЃЛЏH4K16ЫЎЦНЕФНЕЕЭКЭЫЅРЯЯрЙиЁЃВЛТлЪЧЙ§БэДяKAT8ЛЙЪЧИјгшзщЕААзШЅввѕЃЛЏУИ (histone deacetylase, HDAC) вжжЦМСДІРэЃЌОљбгГЄZmpste24ЧУГ§аЁЪѓЕФЪйУќ[53]ЁЃMofЪЧВИШщЖЏЮяЯИАћжаH4K16жївЊЕФзщЕААзввѕЃзЊвЦУИЃЌЫќЖдЮЌГжЛљвђзщЭъећадгазХживЊЕФзїгУЃЌMofЕФМѕЩйЛсбгГйγ-H2AXЮЛЕуЕФаЮГЩКЭDNAЫ№ЩЫгІД№[54,55]ЁЃбаОПЗЂЯжЃЌpreLamin AЕФРлЛ§МѕЩйMofдкКЫЛљжЪжаЕФДцдкЧвЪЙЦфЖЈЮЛДэЮѓЁЃгЩгкMofЖдеаФМDNAаоИДЙ§ГЬжаЕФ53BP1БиВЛПЩЩйЃЌЫљвдprelamin AЕФРлЛ§бгГйСЫDNAЕФЫ№ЩЫаоИДЃЌМгЫйСЫЫЅРЯНјГЬ[53]ЁЃ
ЁЁЁЁВИШщЖЏЮяЕФГСФЌаХЯЂЕїНквђзг (silent information regulator, sirtuin) МвзхЪЧЕфаЭЕФЂѓаЭHDACЁЃsirtuinМвзхга7ИіГЩдБЃЌЪЧЕжПЙЫЅРЯЕФЛљвђМвзхЁЃSIRT6ЪЧвЛжжШЅввѕЃЛЏУИЃЌЫќгыКЫаЁЬхЕФЯрЛЅзїгУЯдзХНЕЕЭСЫH3K9КЭH3K56ЕФввѕЃЛЏЫЎЦНЃЌВЂЧвSIRT6ЪЧзюдчЖЈЮЛдкDSBsЫ№ЩЫЮЛЕуНјаааоИДЕФвђзгжЎвЛ[56]ЁЃGhoshЕШ[57]баОПЗЂЯжЃЌlamin AПЩвдгыSIRT6жБНгЯрЛЅзїгУЃЌВЂЧвЪЧSIRT6ЕФФкдДадМЄЛюМСЁЃLamin AЭЈЙ§ДйНјSIRT6вРРЕЕФDNA-PKcsеаФМЕНШОЩЋжЪЁЂCtIPШЅввѕЃЛЏЁЂКЭPARP1ЕФЕЅADPКЫЬЧКЫЫсЗДгІРДгІЖдDNAЫ№ЩЫЃЌМДе§ГЃЕФLamin AПЩвдДйНјSIRT6дкDNAЫ№ЩЫЩЯЕФШОЩЋжЪЖЈЮЛЃЌДгЖјажњSIRT6НщЕМЕФDNAаоИДЁЃдчРЯЫигыSIRT6ЕФЯрЛЅзїгУИќЧПЃЌЕЋЫќВЂЮДЯдзХдіЧПSIRT6ЛюЖЏЁЃЯрЗДЃЌдчРЯЫиЕФДцдкИФБфСЫSIRT6дкШОЩЋжЪЕФе§ГЃЖЈЮЛКЭвРРЕгкDNAЫ№ЩЫЕФЯТгЮаоИДЪТМўЁЃЫцзХдчРЯЫиКЭSIRT6жЎМфЕФНсКЯдНРДдНЧПЃЌдчРЯЫиНЋSIRT6ЯЕдкКЫЙЧМмЃЌПЩФмЮЃМАDNAЫ№ЩЫЪБЕФSIRT6ЖЏдБЁЃетжжНсКЯПЩФмЛсЪЙУИДгЦфЕзЮяжавўВиЦ№РДЃЌЛђепИФБфЦфЙЙЯѓЃЌЪЙУИЛюадЪмЕНЫ№КІЁЃСэЭтЃЌдчРЯЫигыSIRT6ЕФЯрЛЅзїгУПЩФмЛсДйНјЦфЫћЕААзжЪИДКЯЬхгыSIRT6НсКЯЃЌВЂЯїШѕЦфЛюадЃЌНјвЛВНМгЫйЫЅРЯЕФЗЂЩњЁЃдкHGPSЛМепЕФЯИАћжаЃЌдчРЯЫиЕФДцдкгАЯьMofЕФЖЈЮЛКЭSIRT6ЕФМЄЛюЃЌЭЌЪБдчРЯЫиЮоЗЈааЪЙlamin Aе§ГЃЕФЙІФмЃЌЦЦЛЕзщЕААзШчH4K16ЕФввѕЃЛЏЃЌвдМАSIRT6НщЕМЕФDNAЫ№ЩЫаоИДЪТМўМгЫйЫЅРЯ[57]ЁЃ
ЁЁЁЁ5ЁЂ ЮЪЬтгыеЙЭћ
ЁЁЁЁдкHGPSЛМепЕФЯИАћжаЃЌдчРЯЫиЕМжТЛљвђзщЮШЖЈадЯТНЕЕФЯрЙиЗжзгЛњжЦж№НЅБЛЦЦНтЃЌеыЖдетЗНУцЕФСйДВжЮСЦЕФбаОПвВШЁЕУвЛЖЈГЩЙћЁЃЕЋЪЧЃЌФПЧАЕФжЮСЦЗНАИНідкгкИФЩЦЫЅРЯжЂзДКЭбгГЄЪйУќЗНУцЃЌеце§ИљГ§етИіМВВЁЛЙашИќЩюШыЕФбаОПЁЃСэвЛЗНУцЃЌжзСіЯИАћЕФЮоЯодіжГгыЫЅРЯЯИАћЕФжмЦкзшжЭЪЧЯрЛЅЖдСЂЕФЯжЯѓЃЌШУШЫСЊЯыЕНжзСіЫЅРЯСЦЗЈЕФПЩааадЃЌЖјHGPSЛМепЕФЯИАћзїЮЊбаОПШЫРрЫЅРЯЕФРэЯыФЃаЭЃЌЙигкдчРЯЫиКЭжзСіжЎМфЕФЙиЯЕЃЌНќФъРДв§Ц№СЫШЫУЧЕФЙизЂЁЃВЂЧвгаЮФЯзБЈЕРЃЌдчРЯЫиПЩвдвжжЦжзСіЯИАћЕФЧжЯЎКЭЧЈвЦ[58,59]ЃЌЬсЪОдчРЯЫиПЩФмВЮгыжзСіЕФаХКХЭООЖЃЌВЂЦ№ЕНвЛЖЈвжжЦзїгУЃЌЕЋЪЧОпЬхЕФЗжзгЛњжЦЩаВЛЧхГўЁЃдчРЯЫигыЛљвђзщЮШЖЈадЕФЙиЯЕЩцМАЕНаэЖрЗНУцЃЌЫќУЧЯрЛЅзїгУЯрЛЅСЊЯЕЃЌНјвЛВНбаОПЫќУЧдкдчЫЅЙ§ГЬжаЫљЦ№ЕФзїгУЃЌгажњгкНтПЊдчРЯжЂЕФЩёУиУцЩДЃЌВЂЧвЮЊЬНЫїШЫРре§ГЃЕФЫЅРЯЃЌвдМАдкжзСіЕФЫЅРЯжЮСЦЗНУцгРДСЫаТЕФЛњгігыЬєеНЁЃ
ЁЁЁЁВЮПМЮФЯз
ЁЁЁЁ[1] Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence[J]. Trends Cell Biol, 2018, 28 (6) :436-453
ЁЁЁЁ[2] Kubben N, Misteli T. Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases[J]. Nat Rev Mol Cell Biol, 2017, 18 (10) :595-609
ЁЁЁЁ[3] Dechat T, Pfleghaar K, Sengupta K, et al. Nuclear lamins:major factors in the structural organization and function of the nucleus and chromatin[J]. Genes Dev, 2008, 22 (7) :832-853
ЁЁЁЁ[4] Houben F, Ramaekers FC, Snoeckx LH, et al. Role of nuclear lamina-cytoskeleton interactions in the maintenance of cellular strength[J]. Biochim Biophys Acta, 2007, 1773 (5) :675-686
ЁЁЁЁ[5] Gonzalo S, Kreienkamp R, Askjaer P. Hutchinson-Gilford Progeria Syndrome:A premature aging disease caused by LMNA gene mutations[J]. Ageing Res Rev, 2017, 33:18-29
ЁЁЁЁ[6] Ungricht R, Kutay U. Mechanisms and functions of nuclear envelope remodelling[J]. Nat Rev Mol Cell Biol, 2017, 18 (4) :229-245
ЁЁЁЁ[7] Burke B, Stewart CL. The nuclear lamins:flexibility in function[J]. Nat Rev Mol Cell Biol, 2013, 14 (1) :13-24
ЁЁЁЁ[8] Hennekam RC. Hutchinson-Gilford progeria syndrome:review of the phenotype[J]. Am J Med Genet A, 2006, 140 (23) :2603-2624
ЁЁЁЁ[9] Ahmed MS, Ikram S, Bibi N, et al. Hutchinson-Gilford Progeria Syndrome:A Premature Aging Disease[J]. Mol Neurobiol, 2018, 55 (5) :4417-4427
ЁЁЁЁ[10] Musich PR, Zou Y. Genomic instability and DNA damage responses in progeria arising from defective maturation of prelamin A[J]. Aging (Albany NY) , 2009, 1 (1) :28-37
ЁЁЁЁ[11]дјЬЮ, СѕаТЙт, жмжаОќ.дчРЯжЂ (HGPS) ЕФЗЂВЁЛњжЦгыжЮСЦВпТд[J].ЩњЮяЛЏбЇгыЩњЮяЮяРэНјеЙ (Zeng T, Liu XG, Zhou ZJ.Pathogenesis and treatment strategies of premature aging (HGPS) [J]. Prog Biochem Biophys) , 2007, 34 (7) :687-694
ЁЁЁЁ[12] Cobb AM, Murray TV, Warren DT, et al. Disruption of PCNAlamins A/C interactions by prelamin A induces DNA replication fork stalling[J]. Nucleus, 2016, 7 (5) :498-511
ЁЁЁЁ[13] Hilton BA, Liu J, Cartwright BM, et al. Progerin sequestration of PCNA promotes replication fork collapse and mislocalization of XPA in laminopathy-related progeroid syndromes[J]. FASEB J, 2017, 31 (9) :3882-3893
ЁЁЁЁ[14] Wheaton K, Campuzano D, Ma W, et al. Progerin-Induced Replication Stress Facilitates Premature Senescence in Hutchinson-Gilford Progeria Syndrome[J]. Mol Cell Biol, 2017, 37 (14) . pii:e00659-16
ЁЁЁЁ[15] Kubben N, Zhang W, Wang L, et al. Repression of the Antioxidant NRF2 Pathway in Premature Aging[J]. Cell, 2016, 165 (6) :1361-1374
ЁЁЁЁ[16] Cuadrado A, Rojo AI, Wells G, et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases[J]. Nat Rev Drug Discov, 2019, 18 (4) :295-314
ЁЁЁЁ[17] Liu B, Wang J, Chan KM, et al. Genomic instability in laminopathy-based premature aging[J]. Nat Med, 2005, 11 (7) :780-785
ЁЁЁЁ[18] Liu Y, Liu Y, Yang Z, et al. Cooperative interaction of human XPA stabilizes and enhances specific binding of XPA to DNA damage[J]. Biochemistry, 2005, 44 (19) :7361-7368
ЁЁЁЁ[19] Diner EJ, Vance RE. Taking the STING out of cytosolic DNA sensing[J]. Trends Immunol, 2014, 35 (1) :1-2
ЁЁЁЁ[20] Civril F, Deimling T, de Oliveira Mann CC, et al. Structural mechanism of cytosolic DNA sensing by c GAS[J]. Nature, 2013, 498 (7454) :332-337
ЁЁЁЁ[21] Ablasser A, Goldeck M, Cavlar T, et al. c GAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING[J]. Nature, 2013, 498 (7454) :380-384
ЁЁЁЁ[22] Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol[J]. Nat Immunol, 2013, 14 (1) :19-26
ЁЁЁЁ[23] Hartlova A, Erttmann SF, Raffi FA, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity[J]. Immunity, 2015, 42 (2) :332-343
ЁЁЁЁ[24] Yu Q, Katlinskaya YV, Carbone CJ, et al. DNA-damageinduced type I interferon promotes senescence and inhibits stem cell function[J]. Cell Rep, 2015, 11 (5) :785-797
ЁЁЁЁ[25] Yang H, Wang H, Ren J, et al. c GAS is essential for cellular senescence[J]. Proc Natl Acad Sci U S A, 2017, 114 (23) :E4612-E4620
ЁЁЁЁ[26] Kreienkamp R, Graziano S, Coll-Bonfill N, et al. A cell-intrinsic interferon-like response links replication stress to cellular aging caused by progerin[J]. Cell Rep, 2018, 22 (8) :2006-2015
ЁЁЁЁ[27] Szelag M, Piaszyk-Borychowska A, Plens-Galaska M, et al.Targeted inhibition of STATs and IRFs as a potential treatment strategy in cardiovascular disease[J]. Oncotarget, 2016, 7 (30) :48788-48812
ЁЁЁЁ[28] Fuster JJ, Andres V. Telomere biology and cardiovascular disease[J]. Circ Res, 2006, 99 (11) :1167-1180
ЁЁЁЁ[29] Allsopp RC, Vaziri H, Patterson C, et al. Telomere length predicts replicative capacity of human fibroblasts[J]. Proc Natl Acad Sci U S A, 1992, 89 (21) :10114-10118
ЁЁЁЁ[30] Rodriguez S, Coppede F, Sagelius H, et al. Increased expression of the Hutchinson-Gilford progeria syndrome truncated lamin A transcript during cell aging[J]. Eur J Hum Genet, 2009, 17 (7) :928-937
ЁЁЁЁ[31] Cao K, Graziotto JJ, Blair CD, et al. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in HutchinsonGilford progeria syndrome cells[J]. Sci Transl Med, 2011, 3 (89) :89ra58
ЁЁЁЁ[32] Benson EK, Lee SW, Aaronson SA. Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence[J].J Cell Sci, 2010, 123 (Pt 15) :2605-2612
ЁЁЁЁ[33] Decker ML, Chavez E, Vulto I, et al. Telomere length in Hutchinson-Gilford progeria syndrome[J]. Mech Ageing Dev, 2009, 130 (6) :377-383
ЁЁЁЁ[34] Saha B, Zitnik G, Johnson S, et al. DNA damage accumulation and TRF2 degradation in atypical Werner syndrome fibroblasts with LMNA mutations[J]. Front Genet, 2013, 4:129
ЁЁЁЁ[35] Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1[J].Nature, 2007, 448 (7157) :1068-1071
ЁЁЁЁ[36] Wood AM, Rendtlew DJ, Lucas CA, et al. TRF2 and lamin A/C interact to facilitate the functional organization of chromosome ends[J]. Nat Commun, 2014, 5:5467
ЁЁЁЁ[37] Jeon HJ, Kim YS, Kim JG, et al. Effect of heterochromatin stability on intestinal stem cell aging in Drosophila[J]. Mech Ageing Dev, 2018, 173:50-60
ЁЁЁЁ[38] Heyn H, Moran S, Esteller M. Aberrant DNA methylation profiles in the premature aging disorders Hutchinson-Gilford Progeria and Werner syndrome[J]. Epigenetics, 2013, 8 (1) :28-33
ЁЁЁЁ[39] Lopez-Otin C, Blasco MA, Partridge L, et al. The hallmarks of aging[J]. Cell, 2013, 153 (6) :1194-1217
ЁЁЁЁ[40] Larson AG, Elnatan D, Keenen MM, et al. Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin[J]. Nature, 2017, 547 (7662) :236-240
ЁЁЁЁ[41] Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome[J]. Nat Med, 2005, 11 (4) :440-445
ЁЁЁЁ[42] Pegoraro G, Kubben N, Wickert U, et al. Ageing-related chromatin defects through loss of the NURD complex[J]. Nat Cell Biol, 2009, 11 (10) :1261-1267
ЁЁЁЁ[43] Carrero D, Soria-Valles C, Lopez-Otin C. Hallmarks of progeroid syndromes:lessons from mice and reprogrammed cells[J]. Dis Model Mech, 2016, 9 (7) :719-735
ЁЁЁЁ[44] Jin C, Li J, Green CD, et al. Histone demethylase UTX-1regulates C. elegans life span by targeting the insulin/IGF-1signaling pathway[J]. Cell Metab, 2011, 14 (2) :161-172
ЁЁЁЁ[45] Marullo F, Cesarini E, Antonelli L, et al. Nucleoplasmic Lamin A/C and Polycomb group of proteins:An evolutionarily conserved interplay[J]. Nucleus, 2016, 7 (2) :103-111
ЁЁЁЁ[46] Zhang H, Sun L, Wang K, et al. Loss of H3K9me3 Correlates with ATM Activation and Histone H2AX Phosphorylation Deficiencies in Hutchinson-Gilford Progeria Syndrome[J]. PLoS One, 2016, 11 (12) :e 0167454
ЁЁЁЁ[47] Sun Y, Jiang X, Xu Y, et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60[J].Nat Cell Biol, 2009, 11 (11) :1376-1382
ЁЁЁЁ[48] Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, et al. DNA doublestrand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin[J]. Proc Natl Acad Sci U S A, 2014, 111 (25) :9169-9174
ЁЁЁЁ[49] Harr JC, Gonzalez-Sandoval A, Gasser SM. Histones and histone modifications in perinuclear chromatin anchoring:from yeast to man[J]. EMBO Rep, 2016, 17 (2) :139-155
ЁЁЁЁ[50] Pindyurin AV, Ilyin AA, Ivankin AV, et al. The large fraction of heterochromatin in Drosophila neurons is bound by both B-type lamin and HP1a[J]. Epigenetics Chromatin, 2018, 11 (1) :65
ЁЁЁЁ[51] Liu B, Wang Z, Zhang L, et al. Depleting the methyltransferase Suv39h1 improves DNA repair and extends lifespan in a progeria mouse model[J]. Nat Commun, 2013, 4:1868
ЁЁЁЁ[52] Ziv Y, Bielopolski D, Galanty Y, et al. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM-and KAP-1 dependent pathway[J]. Nat Cell Biol, 2006, 8 (8) :870-876
ЁЁЁЁ[53] Krishnan V, Chow MZ, Wang Z, et al. Histone H4 lysine 16hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice[J]. Proc Natl Acad Sci U S A, 2011, 108 (30) :12325-12330
ЁЁЁЁ[54] Sharma GG, So S, Gupta A, et al. MOF and histone H4acetylation at lysine 16 are critical for DNA damage response and double-strand break repair[J]. Mol Cell Biol, 2010, 30 (14) :3582-3595
ЁЁЁЁ[55] Li X, Corsa CA, Pan PW, et al. MOF and H4 K16 acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1[J]. Mol Cell Biol, 2010, 30 (22) :5335-5347
ЁЁЁЁ[56] Toiber D, Erdel F, Bouazoune K, et al. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling[J]. Mol Cell, 2013, 51 (4) :454-468
ЁЁЁЁ[57] Ghosh S, Liu B, Wang Y, et al. Lamin A is an endogenous SIRT6 activator and promotes SIRT6-mediated DNA repair[J].Cell Rep, 2015, 13 (7) :1396-1406
ЁЁЁЁ[58] Fernandez P, Scaffidi P, Markert E, et al. Transformation resistance in a premature aging disorder identifies a tumorprotective function of BRD4[J]. Cell Rep, 2014, 9 (1) :248-260
ЁЁЁЁ[59] de la Rosa J, Freije JM, Cabanillas R, et al. Prelamin A causes progeria through cell-extrinsic mechanisms and prevents cancer invasion[J]. Nat Commun, 2013, 4:2268