病理学论文


0 引言
非酒精性脂肪性肝病(non-alcoholic fatty liverdis eas e, N A FLD), 是一种以肝脏脂肪浸润、肝细胞炎症坏死为特征的慢性疾病, 可以由单纯性脂肪肝进展至非酒精性脂肪性肝炎(non-alcoholic steatohepatitis, NASH)、肝硬化, 甚至肝细胞肝癌. NAFLD在西方发达国家发病率24%-42%, 而在糖尿病患者中发病率可达70%-80%[1,2]. 2009年Fan等[3]报道上海脂肪肝发病率为17%. 2012年我们研究[4,5]发现, 在上海工作人群中约38.1%的人患有脂肪肝, 其中男性多于女性, 随着年龄增加, 患病率增加, 在上海老年人群中患病率可以达到57.67%. NAFLD已逐渐成为影响人民健康的常见疾病.
目前NAFLD的发病机制仍未明确, 细胞因子及胰岛素抵抗在发病中起了重要作用[6]. 白介素-6(interleukin-6, IL-6)第一个报道可以预测或者导致胰岛素抵抗细胞因子[7]. 一系列的研究发现在血浆和/或组织中的IL-6水平升高会导致胰岛素抵抗[8]. 在肥胖和2型糖尿病患者中, IL-6的水平是升高的[9]. 进行减肥手术(bariatric surgery)的患者血清中IL-6浓度降低, 同时伴随患者的体质量减轻和胰岛素抵抗减轻. 饮食诱导的肥胖小鼠使用IL-6抗体治疗后, 胰岛素的敏感性明显增强, 同时血脂可回归至正常水平. IL-6-/-基因敲除小鼠存在胰岛素抵抗, 而且小鼠成年后表现为肥胖体型, 血中胆固醇和甘油三酯水平较高[10]. 而I L-6+/+转基因小鼠则无胰岛素抵抗, 血中胆固醇和甘油三酯水平较低[11]. 以上研究结果提示,IL-6在胰岛素抵抗和脂代谢中起重要作用.
I L-6属于I L-6细胞因子家族, 可由脂肪细胞、免疫细胞和内皮细胞分泌, 可作为肝脏细胞刺激因子, 诱导各种急性期基因的表达. IL-6通过结合IL-6受体后诱导gp130形成同二聚体发挥作用, 但对于细胞内gp130如何发挥作用的机制仍不确切[12,13]. 我们认为, I L-6参与了NAFLD的胰岛素抵抗及炎症反应过程, 但其是如何通过信号通路来影响脂肪肝的发生和发展仍然不明确, 故进行了本项研究. 本研究首次尝试使用人肝脏L-02细胞进行脂肪肝实验[14,15],期望通过脂肪肝的细胞模型来进一步研究IL-6可能发挥作用的细胞因子信号通路, 为治疗NAFLD提供新的靶点.
NAFLD提供新的靶点.
1 材料和方法
1.1 材料 小牛血清、RPMI 1640培养基(Gibco公司); 亚油酸(sodium oleate)、棕榈酸(sodium pal-mitate)均购自Sigma aldrich公司; TRIzol(Gibco公司); 细胞总RNA提取试剂盒(上海生工生物技术服务有限公司); 实时定量PCR SYBR green PCRmaster mix试剂盒(TaKaRa公司); 细胞蛋白裂解液(RIPA, 上海申能博采); 蛋白质含量检测试剂盒BCA(bicinchoninic acid, PIERCE公司); 蛋白质转移用硝酸纤维膜(Bio-Rad公司); 所有一抗均购自Cell Signaling公司; 所有二抗均购自SantaCruz公司; 蛋白条带显色剂ECL(Amersham);ELISA试剂盒(妙通生物公司); 蛋白芯片杂交标准流程和试剂盒(Full Moon公司). 4 ℃离心机(预冷Fresco 17, Thermo Scientific, USA); 常温离心机(Pico21, Thermo Scientific, USA); 酶标仪(PE Victor, PE, USA); 涡旋振荡器(Vortex 5,QILINBER, China); 摇床(TS-2, Kylin-Bell LabInstruments, China); 芯片小型离心机(ChipMatePMC-082, Tomy, Japan); 芯片扫描仪(GenePix4000B, Axon Instruments, USA); 软件GenePixPro 6.0(Axon Inxtruments, USA); Rotor Gene Q定量PCR仪(Qiagen公司).
1.2 方法
1.2.1 细胞培养及脂肪肝模型构建: 人肝细胞株L-02用含10%小牛血清的RPMI 1640培养基培养(37 ℃、50 mL/L CO2). 将亚油酸和棕榈酸分别以3∶0、2∶1、1∶1、1∶2、0∶3作用于培养的细胞, 24 h后, 形成脂肪浸润的肝脏细胞模型. 应用MTT方法进行脂肪酸的细胞毒性检测. 使用1 mg/L尼罗红进行荧光染色, 在荧光显微镜下(激发光488 nm, 发射光550 nm),运用图像分析系统选择最佳比例浓度的脂肪肝细胞模型.
1.2.2 细胞因子给药: IL-1β、IL-6、IL-17α分别作用于细胞, MTT方法检测细胞毒性发现以上细胞因子对L-02细胞的增殖和凋亡无明显影响, 故选用常规浓度IL-6: 20 ng/mL、IL-1β: 2 ng/mL、IL-17α: 10 ng/mL作用于细胞.
1.2.3 R N A提取-逆转录-实时定量P C R: 应用TRIzol按照细胞总RNA提取试剂盒说明书提取总RNA. 将随机引物及RNA模板在70 ℃变性5 min, 加入逆转录Buffer、dNTP、RNase抑制剂、MMuLV逆转录酶后, 25 ℃温育10 min,42 ℃反应1 h, 70 ℃灭活10 min, 冰浴5 min, 合成cDNA. 实时定量PCR各实验组以20 μL为反应体系, 每20 μL反应体系中加入1 μL cDNA. 实验使用Rotor Gene Q定量PCR仪. 反应采用两步法进行, 反应条件为: 95 ℃ 2 min热启动; 95 ℃ 10 s,60 ℃ 30 s, 共40个循环. 程序设定在每个循环的第二个步骤收集荧光信号, 最后由软件自动生成荧光动力学曲线图, 并据此进行相对定量分析.
1.2.4 Western blot: 将细胞离心沉淀后加入裂解液, 蛋白含量用BCA蛋白质检测试剂盒检测定量. 蛋白质进行变性聚丙烯酰氨凝胶电泳分离后,转移至硝酸纤维膜上. 膜用5%脱脂奶粉室温封闭1 h. 一抗于4 ℃杂交过夜, 二抗于室温杂交1 h, 蛋白条带用ECL显色.
1.2.5 ELISA: 在L-02细胞中按上述方法诱导脂肪肝细胞模型, 收集细胞培养液, 以1000 g离心15 min, 收集上清待测. 根据ELISA试剂盒说明书步骤操作, 采用双抗体夹心ELISA法, 将待测样本或不同浓度标准品加入包被了细胞因子单抗的酶标板中, 并依次加入生物素化的细胞因子抗体和辣根过氧化物酶标记的亲和素反应,加入显色底物显色, 最终加入终止液终止反应,在450 nm处测A值. 以标准物的浓度为横坐标,A值为纵坐标, 计算出标准曲线的直线回归方程式, 将样品的A值代入方程式, 计算出样品浓度,即为样品的实际浓度.
1.2.6 蛋白质芯片: 按照提供的杂交标准流程和试剂盒进行芯片杂交、洗涤与检测. 具体方法如下: 常规裂解蛋白质后, 使用BCA蛋白质检测试剂盒测定蛋白质浓度; 每样本取50 μg蛋白, 每管加入3.0 μL Biotin/DMF, 终体积70 μL, 进行蛋白质样品标记; 芯片封闭; 芯片杂交; 扫描芯片进行检测.
统计学处理 实验结果用SPSS17.0软件进行统计分析, 采用方差分析法比较各组均数之间的差异,P<0.05为差异有统计学意义.
2 结果
2.1 建立L-02细胞脂肪肝细胞模型 MTT试验发现细胞的死亡率与棕榈酸的浓度增加而明显增加, 亚油酸和棕榈酸以2∶1比率, 混合浓度为1.0 mmol/L时细胞毒力最小, 活力最强. 荧光检测发现脂肪在肝脏L-02细胞的聚集率为89.2%±5.4%, 而对照组的脂肪聚集率为5.2%±2.6%(P<0.05)(图1).
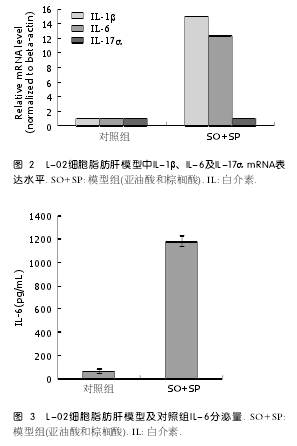
2.2 IL-1β、IL-6、IL-17α在肝脏L-02脂肪肝细胞模型中的mRNA表达水平 实时定量PCR检测的结果可以看到, 与未处理对照组相比, 脂肪肝细胞模型组中IL-1β和IL-6的mRNA水平显著增加,IL-1β升高15.1倍, IL-6升高10.2倍, 而IL-17α表达水平没有明显变化, 所以我们推测炎症因子IL-1β和IL-6在L-02细胞模型脂肪肝的形成中可能发挥作用(P<0.05)(图2).
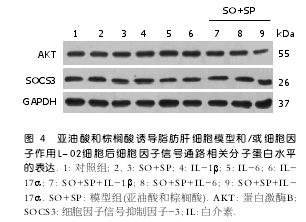
2.3 IL-1β、IL-6、IL-17α在肝脏L-02细胞脂肪肝模型中的分泌水平 与未处理对照组相比,ELISA结果显示, L-02细胞在未接受任何刺激时,就存在IL-6的分泌(71.8 pg/mL), 但模型组细胞分泌量显著增加(1187.8 pg/mL)(P<0.05), 这提示IL-6在L-02细胞脂肪浸润过程中发挥了重要作用(图3). 对于IL-1β和IL-17α, 无论在对照组及脂肪肝细胞模型组的培养液中, ELISA方法都未能检测到其存在, 提示L-02细胞不分泌这两种细胞因子, 因此, 虽然模型组IL-1β mRNA水平有显著增加, 但其可能不被分泌到细胞外, 对于亚油酸和棕榈酸诱导L-02细胞脂肪肝模型的形成并不发挥重要作用.
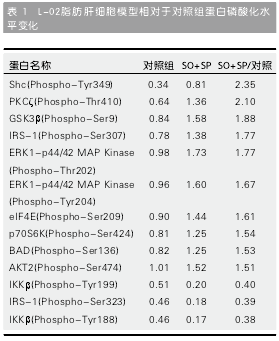
2.4 初步检测IL-1β、IL-6、IL-17α下游信号分子在细胞脂肪肝模型形成前后表达变化 在L-02细胞中按上述方法诱导脂肪肝细胞模型, 同时或单独给予IL-6(或IL-1β、IL-17α). 细胞处理分组为: 对照组、模型组(SO+SP)、单独给予IL-1β组(IL-1β)、单独给予IL-6组(IL-6)、单独给予IL-17α组(IL-17α)、模型组同时给予IL-1β组(IL-1β+SO+SP)、模型组同时给予IL-6组(IL-6+SO+SP)及模型组同时给予IL-17α组(IL-17α+SO+SP). 应用Western blot方法我们初步检测了各组细胞中细胞因子相关信号通路中胰岛素受体底物1(insulin receptor substrate 1,IRS-1)、Grb2相关结合蛋白2(Grb2-associatedbinder 2, Gab2)、a-tubulin、磷脂酰肌醇3-激酶(phosphoinositide-3-kinase, PI3K)、蛋白激酶B(protein kinase B, AKT)及细胞因子信号抑制因子-3(suppressor of cytokine signaling-3, SOCS3)等分子的蛋白表达, 如图4中所示,AKT和SOCS3能够很好表达, 但各组细胞表达量无明显差别.
由于其他蛋白分子抗体较弱, 未能得到结果, 因此, 在后续的实验中利用蛋白质芯片方法进一步检测, 以明确IL-6细胞因子可能的作用机理.
2.5 蛋白芯片检测IL-6下游信号通路分子磷酸化水平变化 我们应用蛋白质芯片技术分别检测了L-02细胞对照组以及脂肪肝模型组中IL-6下游信号通路蛋白分子的磷酸化水平(图5). 对芯片结果进行分析, 结果显示IL-6下游Shc/ERK信号通路(Shc, ERK), mTOR信号通路(p70S6K),PI3K/AKT信号通路(AKT2)均有激活, 而下游NF-κB信号通路可能受到抑制(IKKβ). 同时, 表1结果显示已有文献报道[16-19]过的IL-6能够引起磷酸化的糖原合成酶激酶-3(glycogen synthasekinase-3β, GSK-3β)、蛋白激酶Cζ(protein kinaseCζ, PKCζ)、真核起始因子4E(eukaryotic initia-tion factor 4E, eIF4E)等分子的磷酸化水平也显著上调. 这些结果提示L-02细胞脂肪浸润过程中, IL-6表达量上调并分泌后, 其下游信号通路被广泛激活.
此外有文献报道[20,21], IL-6与肝脏胰岛素抵抗密切相关, 蛋白芯片的结果也显示, 胰岛素受体IRS-1第307位丝氨酸磷酸化水平上调, 而IRS-1第323位丝氨酸磷酸化下调(表1), 这提示在L-02细胞脂肪肝模型中胰岛素信号通路受到了破坏, 因此IL-6也可能通过影响胰岛素信号通路参与细胞脂肪肝模型形成.
3 讨论
目前研究[20,21]认为, 胰岛素抵抗以及机体慢性炎症参与NAFLD的形成. 胰岛素抵抗可以增加游离脂肪酸(free fatty acid, FFA)进入肝脏, 而且刺激肝脏脂肪合成中各种酶的活性, 进一步促进脂质沉积, 也可以诱导氧化应激和刺激肝脏星状细胞增殖分泌细胞外基质促进肝脏纤维化.
而炎症因子可以参与NAFLD肝脏组织细胞的代谢、炎症反应、细胞死亡、再生和纤维化.
上述研究我们发现, 在脂肪酸处理的肝脏L-02细胞中, IL-6在细胞内外表达均明显升高,这说明IL-6在脂代谢中发挥重要的作用. 目前无相关文献报道IL-17α与NAFLD的关系, 我们研究发现IL-17α在体外肝脏细胞表达无变化, 因此IL-17α可能不参与NAFLD形成的机制. 我们研究还发现IL-1β在体外肝脏细胞内mRNA水平表达显著升高, 但是在细胞外并无分泌表达. 然而一些体内研究[22]发现, 人血浆高IL-1β水平可以预测2型糖尿病发生, 合并IL-6升高时更有价值.
脂肪组织中IL-6水平约是血浆中的100倍, 还可以分泌高水平的IL-1β, 诱导炎症反应. 在小鼠脂肪细胞中, 延长IL-1β治疗可以减少胰岛素诱导的葡萄糖摄取[23]. 应用IL-1β受体拮抗剂可以增加脂肪细胞胰岛素敏感性, 致AKT磷酸化的水平增加[24]. 因此, 我们推测, IL-1β在体内可能由脂肪等其他组织自分泌和旁分泌产生, 从而对机体NAFLD的形成发挥作用.
I L-6和其受体结合后需要激活一个含130kDa的信号转导分子gp130才能发挥作用[25]. 与gp130作用后, 低亲和力的IL-6结合位点转变为高亲和力的位点. gp130是IL-6家族分子共享的细胞信号转导受体, 目前研究发现其主要的路径有: (1)活化酪氨酸激酶(janus activated kinase,JAK), 通过STAT3(signal transducers and activa-tors of transcription 3)/SOCS3的酪氨酸磷酸化转导进入核内; (2)激活Ras-MAP(ras-mitogen-activated protein)激酶路径, 通过腺苷酸活化蛋白激酶(AMP-activated protein kinase, AMPK)活化而进入细胞核内, 作用于核内底物NF-IL-6[26].
Giordano等[27]报道, IL-6与gp130结合后还可以激活有丝分裂原蛋白激酶ERK-1和-2(细胞外信号调节酶). 而活化ERK-1和ERK-2需要Src形成Shc-Src同源体. Pelosi等[28]报道在体外肌纤维细胞中IL-6可以通过下调mTOR/p70S6K来影响细胞分化. Chew等[29]报道IL-6通过STAT1和STAT3磷酸化结合至PPARα启动子而抑制PPARα转录, 来调节JAK2/STAT1-3和PI3K/AKT/mTOR通路. 我们研究亦发现IL-6下游Shc/ERK信号通路(Shc, ERK)、mTOR信号通路(p70S6K)和PI3K/AKT信号通路(AKT2)均有激活, 而下游NF-κB信号通路可能受到抑制(IKKβ). 此外, 有文献报道[16-19]IL-6能够引起磷酸化的GSK3β、PKCζ、eIF4E等分子的磷酸化水平也显著上调. 这些结果提示L-02细胞脂肪浸润过程中, IL-6表达量上调并分泌后, 其下游信号通路被广泛激活.
已有文献报道[20,21], IL-6与肝脏胰岛素抵抗密切相关, 蛋白芯片的结果也显示, 胰岛素受体IRS-1第307位丝氨酸磷酸化水平上调, 而IRS-1第323位丝氨酸磷酸化下调, 这提示在L-02细胞脂肪肝模型中胰岛素信号通路受到了破坏, 因此IL-6也可能通过影响胰岛素信号通路参与细胞脂肪肝模型形成.
直接干预IL-6/gp130信号通路是未来治疗疾病的靶目标, 目前已经在寻找sgp130Fc应用于临床试验治疗炎症性疾病[30]. 由于时间精力的限制, 虽然在本实验中发现多个信号通路中蛋白被激活, 但是未能进行进一步验证, 这将在今后实验中继续进行, 以希望找到合适的蛋白靶点应用于治疗NAFLD.

非酒精性脂肪肝(non-alcoholicfattyliverdisease,NAFLD)包括了肝脏损伤的一个广泛的疾病谱。从单纯性脂肪变性到非酒精性脂肪性肝炎(non-alcoholicsteatohepatitis,NASH),随后可进展为肝硬化和终末期肝...

随着IL-34在常见疾病研究中不断展开,使得IL-34日益成为人们研究的热点,因此更加深入的探究IL-34在疾病中的功能和作用机制,将为多种疾病尤其是炎性疾病的治疗提供扎实的理论基础和新的靶点。...
白细胞介素27(IL-27)能对多种类型的免疫细胞发挥调控作用,在抗感染、抗肿瘤免疫和自身免疫性炎症等机体免疫反应中起到重要作用。本文将对IL-27及其与疾病易感性的相关研究进展进行综述。1IL-27的分子结构IL-27是由p28(IL-12的p35相关多肽)和EBI3(EB病毒诱...