生物化学论文

摘 要: 随着分子生物学技术的不断发展和需求的多样化,用于核酸检测的各种PCR衍生技术应运而生。数字PCR是一种单分子水平的大规模分区扩增定量核酸检测技术。该技术以微腔室/微孔或微滴作为PCR反应器,无需校准物和绘制标准曲线即可实现对样品初始浓度的绝对定量,具有高灵敏度、高特异性和高精确度的特点。本文详细介绍了数字PCR的技术发展史、作用原理以及仪器平台类型,系统阐述了数字PCR在转基因检测、疾病诊断、环境及食品监管等方面的应用概况,并对该技术的应用前景进行了展望,以期对未来数字PCR的开发利用提供参考。
关键词: 数字PCR; 单分子; 绝对定量;
Abstract: Various derivative technologies based on PCR for nucleic acid detection have emerged with the continuous development and the diverse needs of molecular biology technology. Digital PCR(dPCR) is a nucleic acid detection method for large scale amplification based on a single molecular template, which runs an individual PCR reaction using chambers/wells or droplets. dPCR can be used for absolute quantification for the initial concentration of samples without calibrator and drawing standard curve, showing the characteristics of high sensitivity, specificity, and accuracy. In this review, we introduce the history of technology development, principle, and instrument platform types of digital PCR in detail. Then, we summarize the application of this technology in GMO quantification, disease diagnosis, environment and food supervision. Finally, we describe the application prospect of dPCR, providing a reference for the development and utilization of this technology in the future.
Keyword: digital PCR; single molecule; absolute quantification;
自1983年Kary Mullis等发明PCR这一革命性技术以来,这种核酸分析方法以其简便、直观、经济、快捷等特性在检测领域一直占据主导地位[1]。随着分子生物学技术的不断发展和需求的多样化,基于普通PCR的各种衍生技术应运而生。PCR技术已不仅仅局限于对靶基因进行体外无限扩增,而是由定性转向了更为精确的定量。1992年,Higuchi等[2]最早提出了实时荧光定量PCR(real time quantitative polymerase chain reaction,qPCR)的设想。qPCR通过在PCR反应体系中加入荧光基团,实时监测整个PCR进程中荧光信号的积累,最后利用标准曲线对未知模板进行定量分析。此后,qPCR被广泛应用于临床诊断、生物育种、食品安全检测和法医鉴定等领域。但qPCR依赖于阈值循环数(Ct值)和校准物,受样品中抑制剂影响较大,无法准确检测低丰度样品,这在一定程度上制约了其使用。数字PCR(digital polymerase chain reaction,dPCR)作为DNA定量的新技术在一定程度上弥补了qPCR的不足,无需校准物和绘制标准曲线即可实现对样品初始浓度的绝对定量,比传统qPCR具有更加出色的灵敏度、特异性和精确性。该技术正在临床诊断、转基因成分定量、单细胞基因表达、环境微生物检测和下一代测序(next-generation sequencing,NGS)文库精确定量和结果验证等方面逐渐发挥重要作用[3,4,5,6,7,8]。

1 、dPCR技术的发展
dPCR从研发起始到广泛应用经历了较长的时间跨度。1992年,Sykes等[9]报道了一种基于极限稀释、PCR和泊松数据统计来定量样本初始目标总数的方法。该研究在检测复杂背景下低丰度重链突变基因IgH时,将待检样品进行了极限稀释使每个反应孔中仅含单个模板分子,PCR扩增结束后统计信号,以此获得准确的起始分子数量。该研究虽未明确提出“数字PCR”的概念,但建立了dPCR的基本实验流程:从极限稀释-PCR-泊松数据统计;确定了dPCR检测中一个极其重要的原则——以“终点信号有无”作为定量方法,形成了dPCR的雏形[10]。1997年,Kalinina等[11]使用纳升体积的石英玻璃毛细管进行单克隆模板PCR扩增,根据荧光探针收集每管内单分子信号,该研究发展了单分子定量技术。1999年,Vogelstein和Kinzler[12]在检测和定量直肠癌患者粪便样品中K-RAS基因突变时,手动将基因组DNA样品稀释并等分至384孔板中,待PCR扩增结束后以野生型或突变型探针杂交,结果显示100多个含有基因组DNA的反应孔中,只有4个反应孔显示基因突变。该研究不仅成功开发了一种癌症诊断方法,而且首次正式提出了“数字PCR”的概念,认为若采用更多孔板可使检测灵敏度更高,指明了dPCR的发展方向。但是在这一时期,dPCR操作复杂,耗费人工,难以大规模开展。2003年,Dressman等[13]设计了一个称之为“BEAMing”的实验方案,以一种类似于流式的磁珠乳液扩增方法,将模板、引物、PCR试剂和磁珠的混合物分散至液滴中,大多数液滴不含或含有1个模板,PCR完成后,扩增产物通过生物素–链霉亲和素连接到磁珠上,随后珠子被打破,探针与扩增产物杂交,通过流式细胞仪读取结果。该技术不仅替代了手工在微孔板中进行样品分散的操作,而且将每个油包水乳化的小液滴变成了PCR反应器,其数量可以达到100,000。该技术建立了基于液滴的数字PCR的工作原理,将数字PCR的研究又推进了一步[14]。虽然研究人员一直致力于对数字PCR的操作简化和检测便捷进行优化,但范围多局限在实验室内进行,商业化进程则是非常缓慢。直到2006年,美国Fluidigm公司研制并推出第一台基于芯片的商业化数字PCR系统;美国Quantalife公司研制出最早的微滴式数字PCR平台。此后,多家国内外公司继续研发推出各种数字PCR平台并投入使用。
2 、dPCR技术的原理及仪器分类
2.1、 dPCR作用原理
dPCR的作用原理是将含有核酸分子的反应体系进行极限稀释,通过控制阀门或微滴生成器分散成为体积可小至皮升级的反应单元,每个反应单元作为一个独立的PCR反应器,其中含有或不含待检靶标分子(DNA或RNA),在PCR扩增结束之后,采集每个反应单元信号,并以终点信号的有无作为判断标准(有荧光信号的液滴判读为“1”,无荧光信号的微滴判读为“0”),最后根据泊松分布(Poisson distribution)原理计算待检靶标分子的浓度或拷贝数(图1)。
2.2 、dPCR数据分析与统计假设
在通常情况下,dPCR每个反应单元中可能包含2个或2个以上的目标分子,因此,在dPCR统计分析中引入了泊松统计方法进行数据的校准。泊松分布是一种离散概率分布,是指在固定的时间或空间内发生给定事件的概率,假定事件以固定的平均速率发生,并与上一次以来的事件无关。泊松概率分布公式如下:
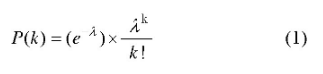
其中是确定观察到目标分子的概率,λ是每个反应单元内平均目标数,是反应单元内实际存在目标分子数。d PCR结果分析是基于以下假设:即目标序列在反应单元内的分布是近乎完美的泊松过程。设定P为阳性反应单元数目,N为总反应单元数目,VP为单个反应单元体积,D为模板稀释因子,每微升样品中DNA拷贝数计算公式见如下[15,16]:
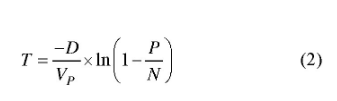
置信区间(confidence interval)是指由样本统计量所构造的总体参数的估计区间。在统计学中,一个概率样本的置信区间是对这个样本的某个总体参数的区间估计。在各平台的仪器设置中,一般选择泊松分布95%的置信区间计算误差线。
2.3、 dPCR仪的分类
目前,dPCR的广泛应用归因于样品分散问题这一关键制约因素的成功解决。纳米加工及微电子技术发展使得高通量分散样品并获得均匀反应单元成为可能。目前市场上使用的数字PCR仪根据样品分散方式可分为两大类:(1)基于微流控芯片数字PCR仪。此类平台又分为封闭式(美国Fluidigm公司BioMark以及美国Formulatrix公司Constellation系统)和开放式(美国ThermoFisher公司Quant Studio)两种平台。封闭式平台是在硅片或石英玻璃上光刻多个微管或微腔室,通过不同的阀门控制溶液的流动,实现样品制备、反应、分离和检测[3];开放式平台是以超高密度亲疏水微孔芯片作为反应载体,芯片表面被处理成疏水,而微孔内部为亲水,这种亲疏水结合的方式可使样品及反应体系轻易进入微孔而不会停留在表面,从而形成密集的独立微反应室并避免了各反应之间的交叉污染[17,18,19]。(2)基于油包水技术的微滴数字PCR平台(美国Bio-Rad公司QX100、QX200和RainDrop系列,中国锐讯生物公司DropX-2000、小海龟BioDigital以及法国Stilla Technologies公司Naica System)。该平台通过液滴发生器将乳化液滴分散成大小均一的液滴反应单元,样品分散过程中的破裂容易造成假阳性,而且分散通道的堵塞问题也是一个关注的重点。相对于基于芯片的数字PCR仪,微滴数字PCR仪价格更为低廉。各种仪器的详细信息见表1所示。
图1 数字PCR的作用原理
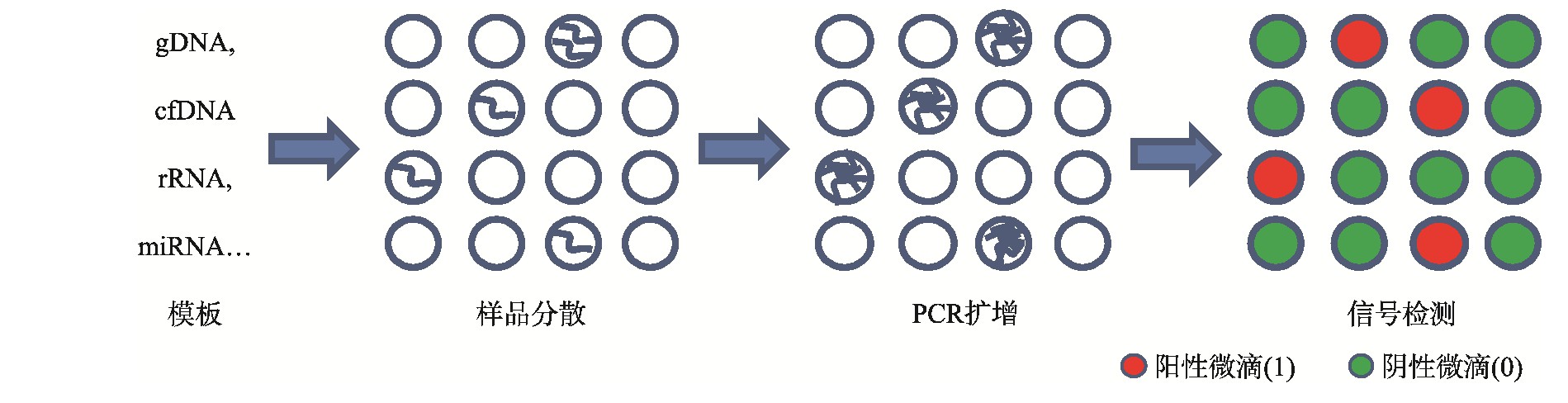
表1 dPCR仪比较
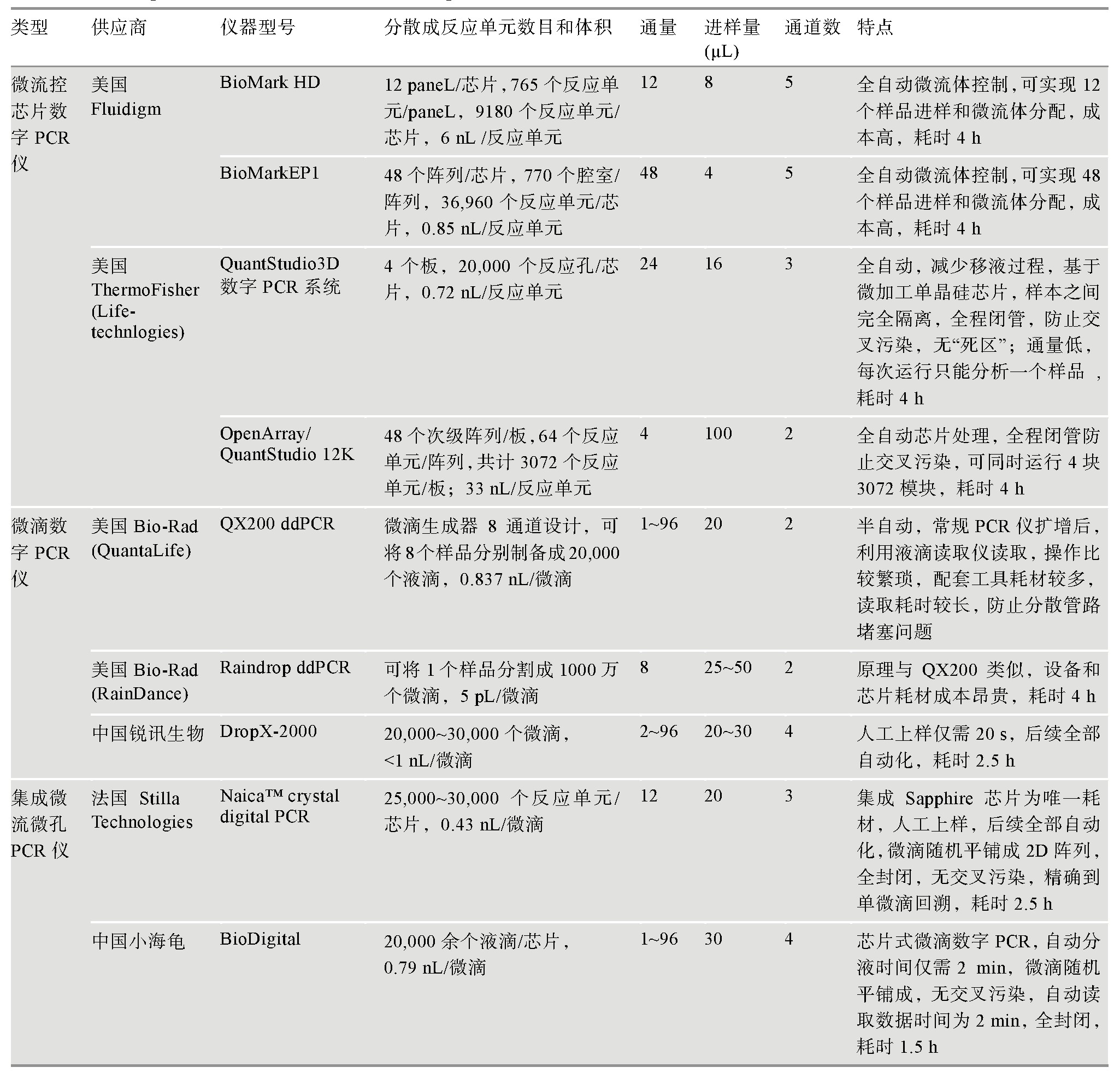
根据参考文献[20,21]汇总修改。
2.3.1 、基于反应室的方法
基于反应室的数字PCR(chamber-based digital PCR,cdPCR)工作流程是将预先混合的PCR反应液通过微流控技术生成乳化液滴并注入预制的固态隔板(室)进行样品分散,然后移至专用热循环仪上进行PCR扩增,读取荧光信号,统计阳性腔室的数目进行分析计算[22]。每种设备所装载反应腔室数目和大小不同,同一设备上各反应腔室大小相同,可确保各腔室之间运行环境一致,cdPCR平台可分散反应单元数量通常低于基于微滴的数字PCR(droplet digital PCR,ddPCR)平台。
2.3.2、 基于微滴的方法
基于微滴的ddPCR工作流程是将PCR预混液与微滴生成油导入微滴生成卡,然后置于微滴生成器生成油包水微滴,每个微滴中含有1个以上或不含目标DNA,移至热循环仪上进行PCR扩增,扩增结束后,利用微滴读数仪读取每个液滴内的终点荧光,最后利用数据分析软件进行分析(https://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulle tin_6311.pdf.)。不同平台所能生成反应单元的数目不同,其具体流程如图2所示。
3、 dPCR技术的应用
dPCR作为继qPCR之后生命科学领域的又一创新,正以其独特的技术优势在转基因植物检测、人类疾病诊断、食品安全、农业与环境监测等领域发挥着重要作用。
3.1、 dPCR在转基因植物检测中的应用
3.1.1 、dPCR对转基因植物拷贝数及合子性的鉴定
生物技术的迅猛发展促进了转基因作物的诞生。在转基因植物研究中,为确保转基因植物的稳定整合和遗传,通常会选择单拷贝的纯合转基因植株作为进一步研究的对象,而且纯合子个体的检测和随后的选择是促进转基因植物从实验室转移到大田的关键。此外,在转基因作物进入市场前,各个国家、组织和地区都制定了明确的政策和法规进行监管,转基因作物中插入的外源基因拷贝数是生物安全评价中一项重要内容[23]。因此,在进行转基因植株检测时会根据需求不同测定其拷贝数与合子性。先前的研究一般采用Southern blot[24,25]、qPCR[26,27,28]和NGS[29]等方法进行检测。但以上检测方法均存在一定程度的缺陷。Southern blot可确定被检转基因植株的拷贝数,也可根据杂交信号亮度差异区分杂合与纯合植株,但模板需求量大而且对DNA质量要求较高,耗时繁琐而且若采用同位素标记探针还会有辐射影响[30]。采用qPCR对于转基因植株的合子性进行检测,不同的实验室获得结果存在争议:或主张该方法无法区分纯合子与杂合子;或可通过精确的扩增条件进行有效区分杂合子[26]。若以qPCR检测转基因植株的拷贝数,对于基因组较大的转基因植株则无法区分一个或两个拷贝,需达到2倍以上的差异[31]。dPCR在转基因植株拷贝数及其杂合子鉴定方面具有模板用量少、节省时间及劳动量和结果更为精准的优势。在玉米(Zea mays L.)中,采用qPCR和ddPCR两种方法均可成功检测出转基因玉米杂合子,但ddPCR因其可直接绝对定量而更为便捷[32]。目前已建立了适用于水稻(Oryza.sativa L.)、柑橘(Citrus sinensis L.)、马铃薯(Solanum tuberosum L.)、玉米(Zea mays L.)、番茄(Solanum lycopersicum L.)和小麦(Triticum aestivum L.)6种转基因作物外源基因拷贝数的ddPCR检测体系[33]。
图2 微滴数字PCR流程示意图
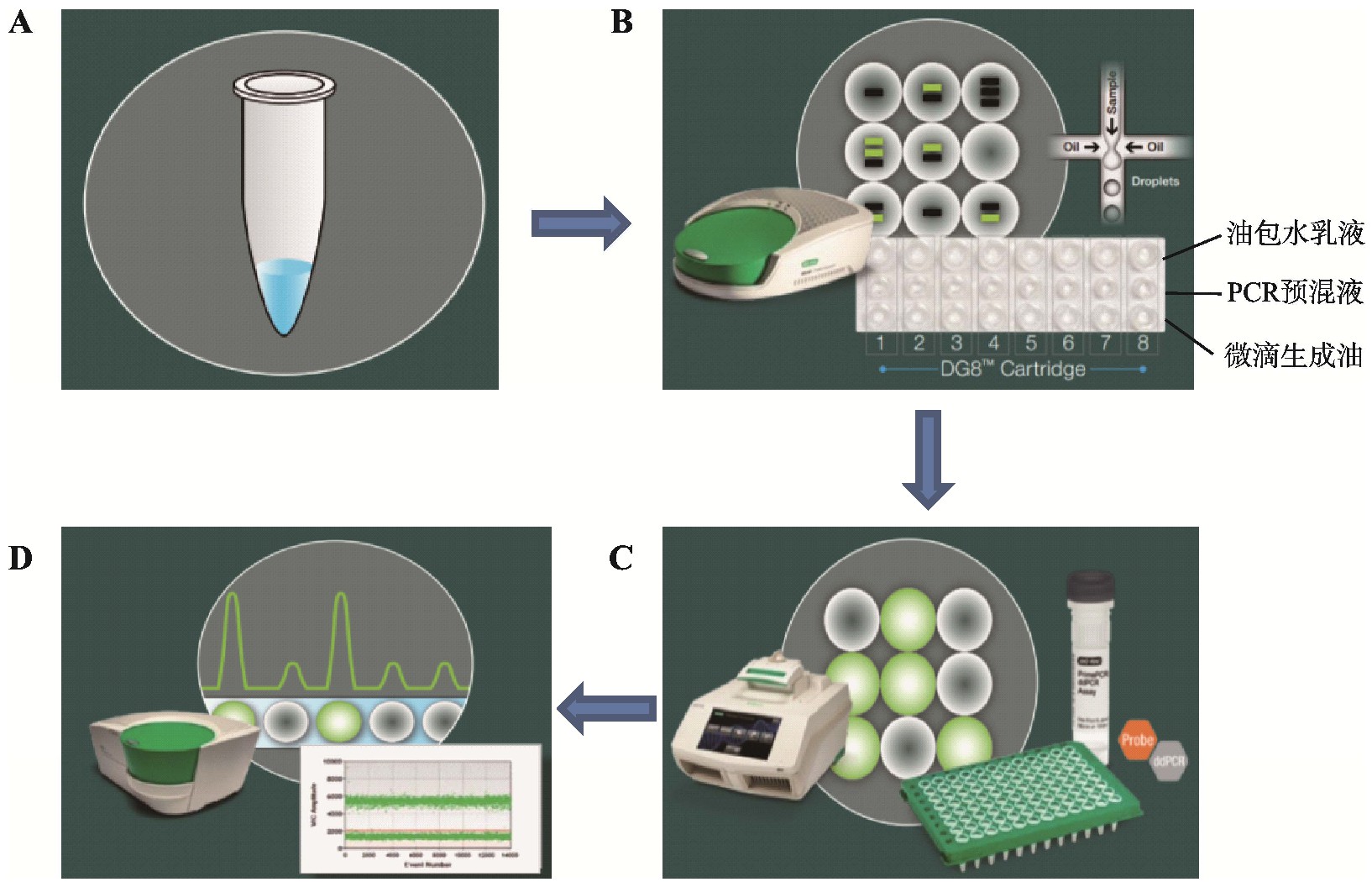
Fig.2 Workflow of droplet digital PCR
A:制备PCR反应液;B:微滴发生器生成油包水乳化液滴;C:在PCR仪进行扩增;D:微滴读取仪读取数据分析结果。
3.1.2、 dPCR对转基因产品成分的检测
自1996年转基因作物商业化生产以来,转基因作物种植面积逐渐增多。据ISAAA(International Service for the Acquisition of Agri-biotech Applications)统计结果,截止到2018年,全球已有70个国家进行了转基因作物应用(种植或进口用作食品/饲料),种植面积已达1.917亿ha[34]。鉴于人们对转基因作物的认知,各国采用了标识制度对转基因产品进行监管,并根据各自需求制定了相应的标识阈值,因此需要相应的技术方法对转基因产品组分进行精确定量[35,36,37]。目前已发布的转基因作物检测标准(物种、元件和事件特异性)多是基于Taqman水解探针的qPCR方法[38,39]。随着技术进步和获批转基因作物数量的增加,复合性状转基因作物已然占据市场主导地位,而基因编辑等新育种技术所培育的品种正推向市场,继续完全使用qPCR方法进行转基因产品成分检测则面临巨大挑战:(1)每种组分的qPCR定量都需要相应的校准品和高质量的标准曲线;(2)检测和定量含有多个转化事件的样品时(如评估食品或饲料标签),总体相对标准偏差(relative standard deviation,RSD)等于所有转化事件定量分析结果的总和,难以在含量临近检测限(limit of detection,LOD)时进行精准定量;(3)抑制剂对校准品和待检样品扩增效率影响较大,易导致定量结果有所偏差;(4)校准品制备繁琐且价格昂贵,增大了检测成本。dPCR可在一定程度上解决使用qPCR检测所存在的问题。
现有qPCR检测标准可以直接转化为dPCR,多个实验室已开始测试并验证了dPCR在转基因生物检测方面的潜力。我国已制定了转基因产品数字PCR检测方法的标准。利用cdPCR方法对转基因玉米MON810有证标准物质进行DNA拷贝数定量分析,获得了与以质粒DNA作为校准品的qPCR方法一致的检测结果,确定了以cdPCR方法检测转基因作物的检测限(limit of detection,LOD)和定量限(limit of quantitation,LOQ)[8,40]。多个研究团队对ddPCR方法中的相关参数(引物浓度、反应单元体积、退火/延伸温度和液滴分离等)进行了优化评估,建立了适用于标准物质和常规检测样品的ddPCR定量分析方法[41,42,43]。Morisset等[44]利用ddPCR对常规的食品和饲料样品中的转基因成分进行了定量适用性分析,与已验证的qPCR和cdPCR相比更为精确和经济有效。
目前市场上推广的dPCR仪多数配有两种荧光通道。从经济效益的角度考虑,多重dPCR具有更好的市场前景。与双重qPCR类似,目前已报道的几种方法可同时定量一个靶标基因和一个内参基因[44,45,46]。利用双通道(例如FAM/VIC)分析3个靶标的检测体系也已建立[47,48,49]。此外,通过同一通道内探针浓度差异(如100 nmol/L和300 nmol/L)进行多重检测也是可选方案。多重dPCR理论上为在一个荧光通道中同时测量某一物种的多个欧盟授权事件和在另一个荧光通道中测量物种内参基因提供支持。Dobnik等[50]利用ddPCR建立了针对欧盟授权的12种转基因玉米品系转基因成分多重定量分析方法。Dobnik等[51]应用多重ddPCR对欧盟授权转基因大豆进行了检测并获得了实验室间联合验证。以上方案均是针对同一物种多个转化事件,并未将具有复合性状转化事件考虑在内。
3.2、 dPCR在人类疾病诊断中的应用
随着分子生物学技术的迅速发展和人类全基因测序的完成,研究发现某些肿瘤是由基因片段缺失、插入、突变或表观遗传修饰造成,而这种变异仅发在肿瘤细胞上,因此可将这种变异作为鉴定肿瘤发生的标记物。当人有一个或多个特定基因组区域出现缺失/复制时,就会发生拷贝数的变异(copy number variations,CNVs),这种CNVs可以自然发生,但在人类中可用作疾病状态的指示。因此,dPCR可应用于肿瘤早期诊断、产前筛查及感染性疾病的判定[52,53]。在对唐氏综合征(downs syndrome,DS)和乳腺癌(breast cancer)等由非整倍体改变而致病的疾病诊断中,使用dPCR方法可更为精确地检测到微小的倍数变化[54]。镰状细胞贫血(sickle cell anemia)是一种由HBB基因突变引起的常染色体显性遗传病,可导致高达5%的胎儿死亡率及4.62%的孕妇死亡率,精准有效的无创产前诊断是预防镰状细胞贫血患儿出生的有效方法。采用dPCR方法对孕妇血浆中胎儿游离DNA(cff-DNA)进行无创产前检测,可确定87%的男性胎儿(n=45)和75%的女性胎儿(n=20)中HBB基因的突变状态;当cff-DNA浓度大于7%时,可以100%的检测出HBB基因突变[55]。此外,dPCR已被成功应用于对病毒载量的测定,从而辅助对疾病进行诊断和检测疗效[56,57]。
3.3 、dPCR在微生物学领域的应用
微生物学的检测对象包括细菌、真菌、病毒和寄生虫等。一些食源性细菌如蜡样芽孢杆菌(Bacillus cereus)、金黄色葡萄球菌(Staphylococcus aureus)、产气荚膜梭菌(Clostridium perfringens)等和真菌如曲霉(Aspergillus)、镰刀菌(Fusarium)、青霉(Penicillium spp.)等在繁殖后会产生毒素,即使经过诸如加热、辐射或高压等灭杀处理后仍持续存在(如镰刀菌产生的曲霉烯真菌毒素、金黄色葡萄球菌产生的肠毒素和蜡样芽孢杆菌产生的呕吐毒素)[58,59,60]。人们一般使用微生物计数的方法来粗略评估。由于并非所有菌株都产生毒素,所以微生物数量与毒素的产生并不严格相关,此外毒素的产生还会因菌株和环境条件而存在差异。一些食源性病原体(细菌和真菌)产生的毒素DNA在经过如上灭杀后仍然存在。因此,基于DNA的dPCR检测和定量方法可用来评估潜在的毒素。
甲型肝炎病毒(hepatitis A virus,HAV)和诺如病毒(norovirus,NoV)是食源性人类病毒疾病的重要病原体,二者均为RNA病毒,现有常规方法还无法培养此类病毒,检测方法主要依赖于RT-PCR和qPCR。理论上,dPCR可对任何病毒进行定量,病毒载量的测定也是评估与某一食品相关的风险并在技术或流行病学层面验证预防措施有效性的先决条件。Coudray-Meunier等[61]采用dPCR和qPCR两种方法对莴苣(Lactuca sativa L.)和水样中的甲型肝炎病毒和诺如病毒进行了定量。此外,有研究报道dPCR还可用于检测和鉴定人体血液中的寄生虫[62]。
3.4、 dPCR在农业和环境监测领域的应用
农业和环境监测两个领域的共同点是都包括广泛的目标生物体和复杂基质,其中抑制物的存在对监测则是一个巨大的挑战[63,64]。随着监管机构将风险评估的标准由定性检测改为定量检测,对动植物病原体的定量变得愈发重要。早期多采用qPCR方法对粪便污染指示细菌、植物、动物、人类病原体和入侵物种进行检测,从而实现对水质监测和微生物来源的识别[63,65,66,67]。由于植物材料、土壤和废水均是具有高抑制剂的基质,所以通过qPCR进行检测和定量植物病原体的可能性极低,而且难以进行多重PCR检测,从而极易导致检测分析结果产生偏差。dPCR可解决以上存在问题,实践证明了其在粪便来源鉴定、检测水样致病性和物种入侵方面的适用性[44,65,68,69]。
3.5 、dPCR在基因编辑领域的应用
基因编辑是近年来一个重大的技术突破,该技术已经广泛应用于基因功能研究、人类疾病治疗、物种改良等多个方面。定性及精确定量评估基因编辑实验是否成功仍是一大难点。在已知编辑背景信息的情况下,基因编辑频率数字PCR(gene-editing frequency digital PCR,GEF-dPCR)利用两个不同标记的探针,同时对给定样本中编辑的和野生型等位基因进行定量,该方法可与定点测序结果相互验证[70]。dPCR最早是应用在对人类细胞系基因组编辑效率进行检测[71,72];在植物上,首先通过dPCR对CRISPR转基因植株进行高通量筛选,然后根据筛选结果,用限制性内切酶消化/PCR扩增技术对检测数值相对较高的植株进一步分析[73,74]。由于基因编辑作物已经推向市场,欧盟将基因编辑作物监管等同于先前转基因作物[75,76,77],对其检测可借鉴dPCR对编辑效率检测的经验,以实现对基因编辑转基因作物含量的定量检测。
4 、结语与展望
相对于此前的核酸定量方法,dPCR达到了前所未有的灵敏度、特异性和精确性,尤其在复杂基质及痕量样品检测方面具有独特优势,其为分子生物学、医学、微生物和环境科学等领域的研究提供了全新的技术手段和思路。采用dPCR对早期世代转基因植株外源基因拷贝数及合子性进行鉴定,为转基因优异转化体的创制带来了极大便利,缩短了筛选进程,提高了筛选效率。此外,dPCR技术为多个专业领域检测限和阈值设定提供了新的度量标尺。对低含量转基因样品的精准定量检测可为我国转基因产品检测标准的制定及相关法规的出台提供理论与技术支持。
dPCR技术还可与NGS、质谱等多种技术相结合。传统PCR方法虽可完成对靶标序列的富集,但其难以达到与现有测序仪器处理量相匹配的水平,ddPCR因其高度的特异性和敏感性,可实现对复杂目标序列的大规模平行单重扩增,通过与NGS技术结合,即可对人类基因组的特定位点进行定点测序,最大限度的提高NGS基于群体的遗传变异研究的效率,可帮助研究人员探索复杂的遗传景观,发现并验证新的疾病关联,预期可开启一个新的分子诊断时代。因此,通过dPCR技术的不断发展完善,其适用的范围将逐渐扩大,与其他检测技术的结合将更为紧密,期望其在基础研究和应用方面发挥更大的作用。
虽然dPCR技术具有强大的技术优势,但该技术的广泛推广仍存在一定的限制因素:(1)分散技术,基于液滴和腔室的dPCR系统都会受到生成与被接收进入到分析统计中反应单元数目的影响。分散生成的数目越多,检测的灵敏度越高,但大多数分散技术都有一定的“死区”,加载到腔室/液滴发生器中的总样品会有少部分残留,从而形成“死区”,这部分体积的加大将会影响对定量的判断,增加相对标准偏差,特别对极低丰度目标影响更大;(2)微滴的准确区分,阳性/阴性信号簇正确清晰的聚集读取对于实验结果至关重要。需不断优化反应体系,调整目标浓度,以期获得标准偏差允许范围内的正确结果;(3)成本,目前市场上dPCR仪器、所需试剂耗材的成本均高于qPCR,有些都为仪器试剂配套使用,这在一定程度上提高了不可控的成本支出,但时间成本的节省可有所弥补;(4)技术更新,dPCR的整个实验流程,从制样–分样–PCR扩增–数据分析的每一步都需要技术的精进与使用人员的不断学习。
参考文献
[1] Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT,Erlich HA, Arnheim N. Enzymatic amplification of betaglobin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science, 1985, 230(4732):1350–1354.
[2] Higuchi R, Dollinger G, Walsh PS, Griffith R. Simultaneous amplification and detection of specific DNA sequences. Biotechnology(NY), 1992, 10(4):413–417.
[3] Ottesen EA, Hong JW, Quake SR, Leadbetter JR. Microfluidic digital PCR enables multigene analysis of individual environmental bacteria. Science, 2006, 314(5804):1464–1467.
[4] White RA, Blainey PC, Fan HC, Quake SR. Digital PCR provides sensitive and absolute calibration for high throughput sequencing. BMC Genomics, 2009, 10:116.
[5] Kim H, Bartsch MS, Renzi RF, He J, Van de Vreugde JL,Claudnic MR, Patel KD. Automated digital microfluidic sample preparation for next-generation DNA sequencing.J Lab Autom, 2011, 16(6):405–414.
[6] Chan M, Chan MW, Loh TW, Law HY, Yoon CS, Than SS,Chua JM, Wong CY, Yong WS, Yap YS, Ho GH, Ang P,Lee ASG. Evaluation of nanofluidics technology for high-throughput SNP genotyping in a clinical setting. J Mol Diagn, 2011, 13(3):305–312.
[7] Spurgeon SL, Jones RC, Ramakrishnan R. High throughput gene expression measurement with real time PCR in a microfluidic dynamic array. PLoS One, 2008, 3(2):e1662
[8] Corbisier P, Bhat S, Partis L, Xie VRD, Emslie KR.Absolute quantification of genetically modified MON810maize(Zea mays L.)by digital polymerase chain reaction.Anal Bioanal Chem, 2010, 396(6):2143–2150.
[9] Sykes PJ, Neoh SH, Brisco MJ, Hughes E, Condon J,Morley AA. Quantitation of targets for PCR by use of limiting dilution. Biotechniques, 1992, 13(3):444–449.
[10] Li CY. Principle and application of digital PCR. Biotech World, 2014, 11:10–13.李春勇.数字PCR技术原理及应用.生物技术世界,2014, 11:10–13.
[11] Kalinina O, Lebedeva I, Brown J, Silver J. Nanoliter scale PCR with TaqMan detection. Nucleic Acids Res, 1997,25(10):1999–2004.
[12] Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci USA, 1999, 96(16):9236–9241.
[13] Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci USA, 2003, 100(15):8817–8822.
[14] Perkel JM. Life science technologies:The digital PCR revolution. Science, 2014, 344(6180):212–214.
[15] Dube S, Qin J, Ramakrishnan R. Mathematical analysis of copy number variation in a DNA sample using digital PCR on a nanofluidic device. PLoS One, 2008, 3(8):e2876.
[16] Dong LH, Meng Y, Sui ZW, Wang J, Wu LQ, Fu BQ.Comparison of four digital PCR platforms for accurate quantification of DNA copy number of a certified plasmid DNA reference material. Sci Rep, 2015, 5:13174.
[17] Basu AS. Digital assays part I:partitioning statistics and digital PCR. SLAS Technol, 2017, 22(4):369–386.
[18] Hu JY, Jiang Y, Yang LT. Quantification of genetically modified maize(Zea mays)MON863 by QuantStudioTM3D digital PCR. J Agric Biotech, 2016, 24(8):1216–1224.胡佳莹,姜羽,杨立桃.利用QuantStudioTM 3D数字PCR分析转基因玉米MON863含量.农业生物技术学报, 2016, 24(8):1216–1224.
[19] Morrison T, Hurley J, Garcia J, Yoder K, Katz A, Roberts D, Cho J, Kanigan T, Ilyin SE, Horowitz D, Dixon JM,Brenan CJ. Nanoliter high throughput quantitative PCR.Nucleic Acids Res, 2006, 34(18):e123.
[20] Demeke T, Dobnik D. Critical assessment of digital PCR for the detection and quantification of genetically modified organisms. Anal Bioanal Chem, 2018, 410(17):4039–4050.
[21] Baker M. Digital PCR hits its stride. Nat Methods, 2012,9(6):541–544.
[22] Liao PY, Huang YY. Digital PCR:endless frontier of'Divide and Conquer'. Micromachines, 2017, 8(8):231.
[23] General Office of the Ministry of Agriculture. Guidelines for the safety assessment of agricultural genetically modified organisms. 2017.农业部办公厅.农业转基因生物(植物、动物、动物用微生物)安全评价指南. 2017.
[24] Hua ZH, Zhu XF, Lin HS, Gao ZY, Qian Q, Yan MX,Huang DN. Studies of the integration and expression of exogenes in transgenic rice obtained via particle bombardment transformation. Acta Genetica Sinica, 2001,28(11):1012–1018.华志华朱雪峰,林鸿生,高振宇,钱前,颜美仙,黄大年.基因枪转化获得的转基因水稻、中外源基因整合与表达规律研究.遗传学报, 2001, 28(11):1012–1018.
[25] Sridevi G, Sabapathi N, Meena P, Nandakumar R, Samiyappan R, Muthukrishnan S, Veluthambi K. Transgenic indica rice variety Pusa Basmati 1 constitutively expressing a rice chitinase gene exhibits enhanced resistance to Rhizoctonia solani. J Plant Biochem Biot, 2003, 12(2):93–101.
[26] Bubner B, Baldwin IT. Use of real-time PCR for determining copy number and zygosity in transgenic plants.Plant Cell Rep, 2004, 23(5):263–271.
[27] Prior FA, Tackaberry ES, Aubin RA, Casley WL. Accurate determination of zygosity in transgenic rice by real-time PCR does not require standard curves or efficiency correction. Transgenic Res, 2006, 15(2):261–265.
[28] Mieog JC, Howitt CA, Ral JP. Fast-tracking development of homozygous transgenic cereal lines using a simple and highly flexible real-time PCR assay. BMC Plant Biol,2013, 13:71.
[29] Fritsch L, Fischer R, Wambach C, Dudek M, Schillberg S,Schr?per F. Next-generation sequencing is a robust strategy for the high-throughput detection of zygosity in transgenic maize. Transgenic Res, 2015, 24(4):615–623.
[30] Passricha N, Saifi S, Khatodia S, Tuteja N. Assessing zygosity in progeny of transgenic plants:current methods and perspectives. J Biol Methods, 2016, 3(3):e46.
[31] Bubner B, Gase K, Baldwin IT. Two-fold differences are the detection limit for determining transgene copy numbers in plants by real-time PCR. BMC Biotechnol,2004, 4:14.
[32] Xu XL, Peng C, Wang XF, Chen XY, Wang Q, Xu JF.Comparison of droplet digital PCR with quantitative real-time PCR for determination of zygosity in transgenic maize. Transgenic Res, 2016, 25(6):855–864.
[33] Collier R, Dasgupta K, Xing YP, Hernandez BT, Shao M,Rohozinski D, Kovak E, Lin J, de Oliveira MLP, Stover E,McCue KF, Harmon FG, Blechl A, Thomson JG,Thilmony R. Accurate measurement of transgene copy number in crop plants using droplet digital PCR. Plant J,2017, 90(5):1014–1025.
[34] Global status of commercialized biotech/gm crops in 2018.Biotech crop adoption surges as economic benefits accumulate 23 years. ISAAA, brief 54.
[35] Gruère GP, Rao SR. A review of international labeling policies of genetically modified food to evaluate india's proposed rule. AgBioForum, 2007, 10(1):51–64.
[36] Kamle S, Ali S. Genetically modified crops:Detection strategies and biosafety issues. Gene, 2013, 522(2):123–132.
[37] Milavec M, Dobnik D, Yang LT, Zhang DB, Gruden K, Zel J. GMO quantification:valuable experience and insights for the future. Anal Bioanal Chem, 2014, 406(26):6485–6497.
[38] Scholtens IM, Kok EJ, Hougs L, Molenaar B, Thissen JT,van der Voet H. Increased efficacy for in-house validation of real-time PCR GMO detection methods. Anal Bioanal Chem, 2010, 396(6):2213–2227.
[39] Berdal KG, Holst-jensen A. Roundup Ready?soybean event-specific real-time quantitative PCR assay and estimation of the practical detection and quantification limits in GMO analyses. Eur Food Res Technol, 2001, 213:432–438.
[40] Burns MJ, Burrell AM, Foy CA. The applicability of digital PCR for the assessment of detection limits in GMO analysis. Eur Food Res Technol, 2010, 231(3):353–362.
[41] Gerdes L, Busch U, Pecoraro S. Parallelised real-time PCR for identification of maize GMO events. Eur Food Res Technol, 2012, 234(2):315–322.
[42] K?ppel R, Bucher T. Rapid establishment of droplet digital PCR for quantitative GMO analysis. Eur Food Res Technol, 2015, 241(3):427–439
[43] Fu HB, Yan CJ, Li S, Liu XC, Zhang M. Application of digital PCR technology in detection of genetically modified components. Liaoning Agric Sci, 2017,(1):50–53.付海滨,闫超杰,李姝,刘晓超,张敏.数字PCR技术在转基因成分检测中的应用.辽宁农业科学, 2017,(1):50–53.
[44] Morisset D,?tebih D, Milavec M, Gruden K,?el J.Quantitative analysis of food and feed samples with droplet digital PCR. PLoS One, 2013, 8(5):e62583.
[45] Gerdes L, Iwobi A, Busch U, Pecoraro S. Optimization of digital droplet polymerase chain reaction for quantification of genetically modified organisms. Biomol Detect Quantif, 2016, 7:9–20.
[46] Félix-Urquídez D, Pérez-Urquiza M, Valdez Torres JB,León-Félix J, García-Estrada R, Acatzi-Silva A. Development, optimization, and evaluation of a duplex droplet digital PCR assay to quantify the T-nos/hmg copy number ratio in genetically modified maize. Anal Chem, 2016,88(1):812–819.
[47] Dobnik D,?tebih D, Blejec A, Morisset D,?el J.Multiplex quantification of four DNA targets in one reaction with Bio-Rad droplet digital PCR system for GMO detection. Sci Rep, 2016, 6:35451.
[48] Whale AS, Huggett JF, Tzonev S. Fundamentals of multiplexing with digital PCR. Biomol Detect Quantif,2016, 10:15–23.
[49] Pretto D, Maar D, Yrigollen CM, Regan J, Tassone F.Screening newborn blood spots for 22q11.2 deletion syndrome using multiplex droplet digital PCR. Clin Chem,2015, 61(1):182–190.
[50] Dobnik D, Spilsberg B, Bogo?alec Ko?ir AB, Holst-Jensen A,?el J. Multiplex quantification of 12 European Union authorized genetically modified maize lines with droplet digital polymerase chain reaction. Anal Chem, 2015,87(16):8218–8226.
[51] Ko?ir AB, Spilsberg B, Holst-Jensen A,?el J, Dobnik D.Development and inter-laboratory assessment of droplet digital PCR assays for multiplex quantification of 15genetically modified soybean lines. Sci Rep, 2017, 7(1):8601.
[52] Zimmermann BG, Grill S, Holzgreve W, Zhong XY,Jackson LG, Hahn S. Digital PCR:a powerful new tool for noninvasive prenatal diagnosis? Prenat Diagn, 2008,28(12):1087–1093.
[53] Fan MHC, Blumenfeld YJ, El-Sayed YY, Chueh J, Quake SR. Microfluidic digital PCR enables rapid prenatal diagnosis of fetal aneuploidy. Am J Obstet Gynecol, 2009,200(5):543.e1–547.e7.
[54] Usher CL, McCarroll SA. Complex and multi-allelic copy number variation in human disease. Brief Funct Genomics,2015, 14(5):329–338.
[55] Barrett AN, McDonnell TC, Chan KC, Chitty LS. Digital PCR analysis of maternal plasma for noninvasive detection of sickle cell anemia. Clin Chem, 2012, 58(6):1026–1032.
[56] Trypsteen W, Kiselinova M, Vandekerckhove L, De Spiegelaere W. Diagnostic utility of droplet digital PCR for HIV reservoir quantification. J Virus Erad, 2016, 2(3):162–169.
[57] Sedlak RH, Jerome KR. Viral diagnostics in the era of digital polymerase chain reaction. Diagn Microbiol Infect Dis, 2013, 75(1):1–4.
[58] Hennekinne JA, De Buyser ML, Dragacci S. Staphylococcus aureus and its food poisoning toxins:characterization and outbreak investigation. FEMS Microbiol Rev, 2012,36(4):815–836.
[59] Pinchuk IV, Beswick EJ, Reyes VE. Staphylococcal enterotoxins. Toxins, 2010, 2(8):2177–2197.
[60] Stenfors Arnesen LP, Fagerlund A, Granum PE. From soil to gut:Bacillus cereus and its food poisoning toxins.FEMS Microbiol Rev, 2008, 32(4):579–606.
[61] Coudray-Meunier C, Fraisse A, Martin-Latil S, Guillier L,Delannoy S, Fach P, Perelle S. A comparative study of digital RT-PCR and RT-qPCR for quantification of Hepatitis A virus and Norovirus in lettuce and water samples. Int J Food Microbiol, 2015, 201:17–26.
[62] Wilson M, Glaser KC, Adams-Fish D, Boley M, Mayda M,Molestina RE. Development of droplet digital PCR for the detection of Babesia microti and Babesia duncani. Exp Parasitol, 2015, 149:24–31.
[63] Strand DA, Holst-Jensen A, Viljugrein H, Edvardsen B,Klaveness D, Jussila J, Vralstad T. Detection and quantification of the crayfish plague agent in natural waters:direct monitoring approach for aquatic environments. Dis Aquat Organ, 2011, 95(1):9–17.
[64] Ra?ki N, Dreo T, Gutierrez-Aguirre I, Blejec A, Ravnikar M. Reverse transcriptase droplet digital PCR shows high resilience to PCR inhibitors from plant, soil and water samples. Plant Methods, 2014, 10(1):42.
[65] Doi H, Takahara T, Minamoto T, Matsuhashi S, Uchii K,Yamanaka H. Droplet digital polymerase chain reaction(PCR)outperforms real-time PCR in the detection of environmental DNA from an invasive fish species.Environ Sci Technol, 2015, 49(9):5601–5608.
[66] Masago Y, Konta Y, Kazama S, Inaba M, Imagawa T,Tohma K, Saito M, Suzuki A, Oshitani H, Omura T.Comparative evaluation of real-time PCR methods for human noroviruses in wastewater and human stool. PLoS One, 2016, 11(8):e0160825.
[67] Huang WC, Chou YP, Kao PM, Hsu TK, Su HC, Ho YN,Yang YC, Hsu BM. Nested-PCR and TaqMan real-time quantitative PCR assays for human adenoviruses in environmental waters. Water Sci Technol, 2016, 73(8):1832–1841.
[68] Ra?ki N, Morisset D, Gutierrez-Aguirre I, Ravnikar M.One-step RT-droplet digital PCR:a breakthrough in the quantification of waterborne RNA viruses. Anal Bioanal Chem, 2014, 406(3):661–667.
[69] Cao Y, Raith MR, Griffith JF. Droplet digital PCR for simultaneous quantification of general and humanassociated fecal indicators for water quality assessment.Water Res, 2015, 70:337–349.
[70] Mock U, Hauber I, Fehse B. Digital PCR to assess geneediting frequencies(GEF-dPCR)mediated by designer nucleases. Nat Protoc, 2016, 11(3):598–615.
[71] Findlay SD, Vincent KM, Berman JR, Postovit LM. A digital PCR-based method for efficient and highly specific screening of genome edited cells. PLoS One, 2016, 11(4):e0153901.
[72] Miyaoka Y, Chan AH, Judge LM, Yoo J, Huang M,Nguyen TD, Lizarraga PP, So PL, Conklin BR. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat Methods, 2014, 11(3):291–293.
[73] Gao R, Feyissa BA, Croft M, Hannoufa A. Gene editing by CRISPR/Cas9 in the obligatory outcrossing Medicago sativa. Planta, 2018, 247(4):1043–1050.
[74] Liu CX, Geng LZ, Xu JP. Detection methods of genome editing in plants. Hereditas(Beijing), 2018, 40(12):1075–1091.刘春霞,耿立召,许建平.植物基因组编辑检测方法.遗传, 2018, 40(12):1075–1091.
[75] Waltz E. CRISPR-edited crops free to enter market, skip regulation. Nat Biotechnol, 2016, 34(6):582.
[76] Huang S, Weigel D, Beachy RN, Li J. A proposed regulatory framework for genome-edited crops. Nat Genet,2016, 48(2):109–111.
[77] Waltz E. Gene-edited CRISPR mushroom escapes US regulation. Nature, 2016, 532(7599):293.

自1985年KaryMullis发明PCR技术以来,PCR技术一直是生物医学领域中重要的实验方法。随着分子生物学技术的发展,目前核酸定量的主要方法是实时荧光定量PCR(QuantitativePCR,qPCR)。数字PCR(DigitalPCR,dPCR)是近年来发展起来的新技术,是对传统P...

近年来,分子生物学技术发展迅速,尤其是基于PCR衍生出的一系列技术,因其具有操作简单、快速准确、特异性强且灵敏度高的特点,已被广泛应用于各种检测领域。...