生物化学论文

摘 要: 数字PCR (digital PCR, dPCR)是在普通PCR和定量PCR基础上发展的第三代PCR技术,通过有限稀释和泊松分布统计实现精确的绝对定量检测。相比于前两代PCR技术,dPCR能够实现DNA模板的绝对定量且对PCR抑制剂具有更强的耐受性。目前,该技术在致病菌和病毒、基因突变、甲基化DNA、转基因成分和食品掺假的检测中都得到了广泛的应用。该文介绍了dPCR的原理并综述了其在生物学检测中应用的研究进展,以期为基于dPCR的检测方法的开发和应用提供参考和依据。
关键词 : 数字PCR;生物学检测;绝对定量;
Abstract: Digital PCR(dPCR) is a third-generation PCR technology based on conventional PCR and quantitative PCR, which applies finite dilution and poisson statistics to achieve accurate absolute quantitative detection. Compared to the previous two generations of PCR technology, dPCR can achieve absolute quantification of DNA template and shows better tolerance to PCR inhibitors. At present, dPCR has been widely used in detection of pathogenic bacteria, viruses, genetic mutations, methylated DNA, genetically modified ingredients and food adulteration. This review introduces the principle of dPCR, summarizes its progress in biological detection area, with the aim to provide references for development and further application of dPCR.
Keyword: digital PCR; biological detection; absolute quantitative detection;

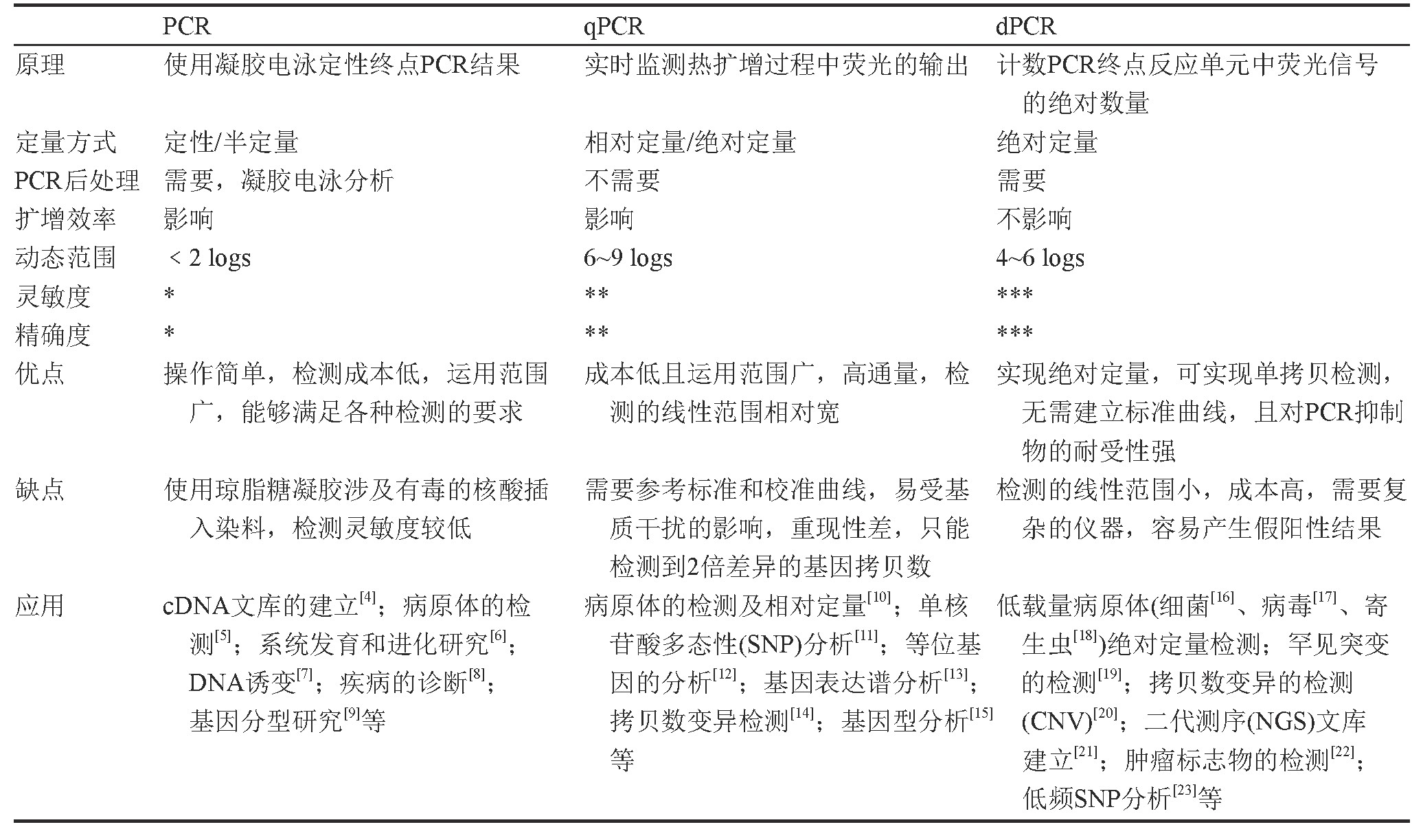
近年来,分子生物学技术发展迅速,尤其是基于PCR衍生出的一系列技术,因其具有操作简单、快速准确、特异性强且灵敏度高的特点,已被广泛应用于各种检测领域。1983年,美国KB Mullis教授发明了PCR技术用于核酸检测,其被称为第一代PCR[1]。随后PCR技术出现井喷式发展,1992年科学家们在第一代PCR技术的基础上引入荧光化学物质,发明了定量PCR技术(quantitative PCR,q PCR)[2]:通过荧光信号的变化对整个PCR过程进行实时监控,最后通过循环阈值(cycle threshold,Ct)和标准曲线对待测样本进行定量检测。数字PCR (digital PCR,d PCR)技术是近年来广泛应用于检测的第三代PCR技术。区别于q PCR依赖校准曲线对靶基因定量的策略,该技术通过将PCR体系分散成无数个小体积的反应单元进行扩增,允许单拷贝DNA的检测,可以实现对核酸的绝对定量。每个小反应单元含有少量或者没有目标序列,这有助于降低样本中的多种抑制剂的干扰和PCR体系中模板之间的竞争效应。自1999年Vogelstein和Kinzler[3]第一次阐述d PCR方法用于基因突变的检测之后,该方法便在各个检测领域陆续得到广泛应用并发展。表1总结了三代PCR技术的特点及其应用范围。
表1 PCR技术的比较
1、 d PCR的原理
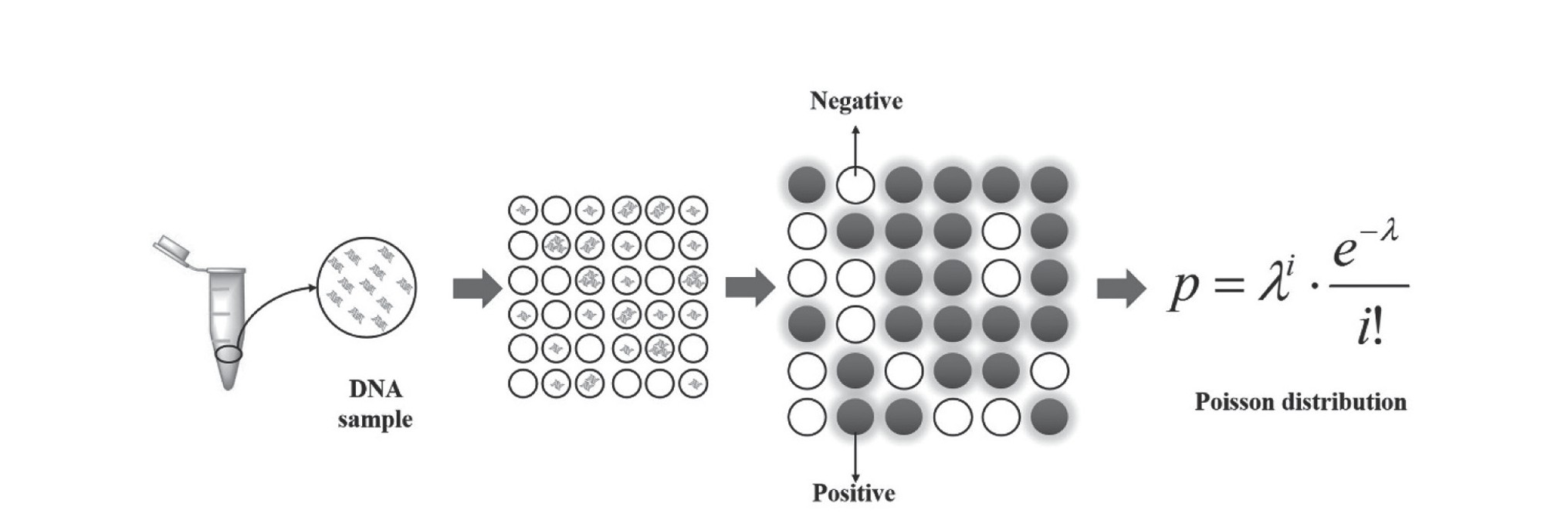
d PCR的原理是通过有限稀释将含有目的DNA的PCR反应体系分散成无数个单一模板的PCR体系进行扩增,再通过统计检测结果及泊松分布校正实现目的DNA的绝对定量(图1)。
d PCR包括三个步骤:分散体系、PCR扩增和信号检测。通过样品的稀释和分散形成分散体系,这一步是限制d PCR发展应用的一个重要因素,其所引起的分散数量和分散体积的不同极大地影响着定量结果的精度与准确性。分散体系的形成增加了靶分子的有效浓度,并在一定程度上对存在干扰的复杂化合物进行了纯化,提高了靶分子和背景之间的比率[24]。基于d PCR统计的定量方式,反应体系的分散数量越多,低浓度靶分子的检测可能性就越大,检测灵敏度就越高,同时多个单一的反应单元也为多目标序列的高通量检测的发展提供了基础。此外,分散体积的不同会对拷贝数结果的测量产生影响,是造成d PCR精度下降的一个潜在因素,且与检测限的大小形成反比关系。
然而,在d PCR发展早期,使用96孔板或384孔板作为单一样品的分散反应载体,其分散程度低、分区数量少,无论是时间和试剂耗材成本,还是检测精确度,都无法满足实际应用的要求。微流体技术和乳液化学的引入为样品分散问题的解决提供了良好的动力。目前已成功应用于商业化的d PCR系统根据分散方式的不同可分为三种主要类型:基于油包水微滴生成技术的微滴式数字PCR (droplet digital PCR,dd PCR)、基于微孔芯片的微孔板数字PCR (micro-chamber digital PCR,md PCR)和基于微流控技术的微流控芯片式数字PCR (microfluidic chip digital PCR,mcd PCR)(表2)。另外,基于水凝胶珠、琼脂糖珠和生物打印技术等样本分散方式的d PCR虽然尚未商业化,但在现有的实验研究中已取得一定进展。微滴式通过油包裹PCR体系形成无数个油包水微滴,每个微滴是一个独立的反应单元,这种特殊的微滴在进行PCR时能够保证完整的形态,不会互相扩散[25],也阻止PCR混合物液滴在热扩增过程的蒸发;微孔芯片式是利用光刻技术在硅基上刻蚀出微孔阵列,再通过表面改性、打磨等操作形成数万个纳升级的表面疏水和孔内壁亲水的微反应室;微流控芯片式则是利用微流控芯片装置使PCR反应体系准确快速地分散于芯片孔中,而每个孔都是一个小反应体系(纳升级)[26]。md PCR操作简单快速,只需一步便可形成均匀分散样品,亲疏水结合的设计使样品不会停留在芯片,从而避免了各反应室的交叉污染,且表面相邻反应之间存在固态隔板,不容易产生假阳性结果,而dd PCR中液滴之间的液体屏障在液滴转移或热扩增过程中存在破碎的风险[27]。dd PCR利用油包水形成液滴,制备难度低、易实现高通量检测,但是在进行分液及转移到反应孔时有部分死体积,会造成检测结果偏差[28];而mcd PCR是通过微流控芯片将反应分配到反应孔中,其对仪器性能要求不高,容易推广使用。相比较于dd PCR,mcd PCR有效避免了其分散通道容易堵塞的问题,但其价格更贵,检测通量低,无法满足大批量样品的同时检测。同时,mcd PCR的分区数量通常少于dd PCR,这导致其检测的动态范围相对更低[29]。进行PCR扩增时,微滴式是通过将分散好的PCR体系移入到96孔板中,然后在普通的PCR仪上进行扩增即可,而芯片式则需要通过特定的仪器进行扩增。
图1 数字PCR的原理
关于信号检测,目前主要有光电倍增管(photomultiplier,PMT)、硅光电子计数器(multi-pixel photon counter,MPPC)、电荷耦合器件(charge-coupled device,CCD)、互补金属氧化物半导体(complementary metal oxide semiconductor,CMOS)、扫描器等5种高分辨率的图像处理策略[30]。对于不同的分散方式,合适的光学检测系统的选择直接关系到定量检测结果的精度、灵敏度。dd PCR基于液滴的荧光信号计数,需要将96孔板放在液滴读取仪中,适合采用PMT/硅光电子计数器对每个微滴进行信号分析,最后经过软件处理输出拷贝数。PMT技术虽然抗干扰能力强,但一次只能处理一个液滴,不适合于高通量的定量检测,其相应的激光光源和软件也相对昂贵[31]。而基于水凝胶珠、琼脂糖珠、生物打印技术、微孔芯片和微流控芯片等的d PCR一般是通过使用扫描设备,如CCD、CMOS等,对芯片的荧光点信号进行捕捉,然后通过读取荧光信号进行分析,最终得出拷贝数。基于扫描方式的荧光系统可以一次性扫描整个反应平台,但其检测时间却主要取决于定量过程中使用的图像处理算法[30,31]。
d PCR定量计数的方法相较于传统q PCR的定量方法更为简单,无需建立标准曲线和计算反应的扩增效率等繁琐步骤,而是通过直接计数的方式得出检测结果。在进行PCR扩增反应后,将有荧光信号(阳性)的标记“1”,没有荧光信号(阴性)的标记“0”,以此进行信号的统计计算。但在理想条件下一个反应单元最多含有一个目标物,而实际操作中d PCR有阳性信号的反应单元可能不止一个目标物,所以根据阳性信号数量进行统计计算时,所得的最终结果不是真实的目标DNA分子拷贝数存在着一定的可能性,因此需要通过泊松分布概率公式对反应的结果进行校正计算[6,7]。在该公式中,λ表示每个反应单元包含靶DNA分子的平均拷贝数,p表示在一定的λ条件下每个反应单元中含有i拷贝靶DNA分子的概率。当样品DNA稀释倍数m一定时,λ的大小也就一定,即λ=cm,c是目的DNA原始拷贝数量。当i值为0时,即没有阳性信号的微孔的概率p=e-λ,而,n是反应的总微孔数,f是阳性信号的微孔数。从而得到,两边同时取对数可得,经换算得。根据最终检测结果,可以通过阴性反应的数量和稀释倍数实现对目标DNA的绝对定量。
2 、d PCR在生物学检测中的应用
d PCR发展至今已近20年,因其具有检测灵敏度高、特异性强且有效避免PCR抑制剂的影响等优势,在分子检测和疾病诊断等领域都得到了广泛应用。根据Pub Med数据库统计结果,截至2020年6月,与d PCR相关的研究报道有3 679篇,尤其是近几年随着d PCR仪器的不断开发,有大量的研究报道进一步证实了d PCR的优势。表3总结了近年来利用d PCR实现生物学检测的研究。

2.1 、致病菌的检测
致病菌,如产毒素的金黄色葡萄球菌、沙门氏菌、单核增生李斯特菌等广泛滋生于人们食用的水、肉、蛋、米饭等日常食物中,是导致食源性疾病发生的主要原因。通过设计致病菌的靶基因序列的特异性引物和进行扩增是应用PCR技术实现检测的主要原理。目前普通PCR和q PCR在致病菌检测中虽然已经得到了广泛的应用,但是在目标分子含量低、检测基质复杂的情况下,最终的检测结果会有明显偏差,限制了其在致病菌检测领域的应用。d PCR作为第三代PCR技术,在前两代基础上进一步发展,提高了检测的灵敏度和准确性,减少了前期的增菌培养过程,大大缩短了检测分析的时间,而且不需要建立标准曲线即可实现对目标分子的绝对定量检测。Wang等[37]分别采用d PCR和q PCR检测牛奶中的鼠伤寒沙门氏菌,结果显示d PCR在加标牛奶中的检测限相比于q PCR低一个数量级;而且通过评价对PCR抑制物(Ca2+和乙醇)的耐受效果,发现d PCR能耐受更高浓度的抑制物。He等[38]利用dd PCR同时检测大肠杆菌O157:H7和O104:H4,以stx和o抗原特异性基因为靶基因,通过使用Taq Man探针对人工污染的苹果汁进行检测,结果目标菌的检测限均为2 cfu/m L。
表3 数字PCR在生物学检测领域的应用
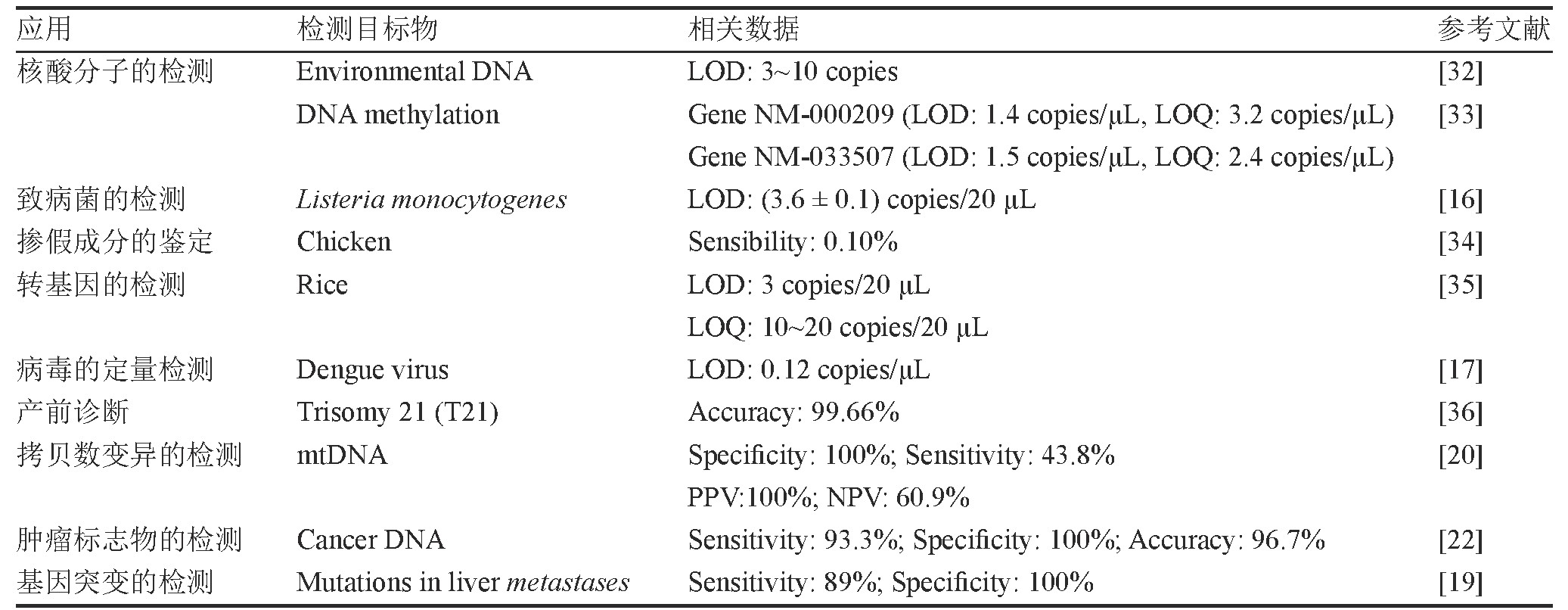
注:LOD,limit of detection;LOQ,limit of quantitative;PPV,positive predictive value;NPV,negative predictive value。
2.2 、病毒的检测
目前病毒检测常用的方法主要是通过免疫学方法检测其蛋白质或通过分子生物学方法检测其特定核酸。d PCR作为最新的分子生物学检测技术,检测时无需建立标准曲线且不受基质效应的影响,适用于病毒分子定量检测尤其是低拷贝病毒的定量检测。自商业化的d PCR产品开发以来,将d PCR应用于病毒的检测,尤其是对病毒载量的量化研究一直是近几年的研究热点,现已有文献报道对新型冠状病毒[39]、流感病毒[40]、肠道病毒[41]、蓝舌病病毒[42]、猪流行性腹泻病毒[43]、感染性脾肾坏死病毒[44]等进行定量分析。其中,为了比较RT-q PCR和RT-d PCR检测病毒的性能,Coudray-Meunier等[45]使用上述两种方法对水和生菜中的诺瓦克病毒和甲型肝炎病毒进行定量检测,通过灵敏度分析发现,RT-d PCR能够检测更低的病毒拷贝数;为了进一步分析对复杂基质的耐受情况,对生菜样本进行加标的检测结果表明RT-d PCR的回收率显着高于RT-q PCR。Pav?i?等[46]利用d PCR直接定量检测病毒,不涉及病毒核酸的提取,结果显示这种直接检测的结果重复性更高且更接近于病毒的真实载量。
2.3、 基因突变的检测
基因突变的检测方法主要是PCR或Sanger测序法,但这两种方法对低含量的基因突变检测灵敏度低[47,48],且难以满足早期突变筛查的需要。d PCR第一次被阐述正是用于基因突变的检测,结果显示d PCR对基因突变的分析灵敏度高,且可以对复杂组织进行检测[3]。人类在患胰腺癌和结肠癌的早期,通常会发生12号染色体上KRAS基因的突变[49]。传统的组织活检是从手术中取样,取样风险高,且对取样的次数也有一定的限制,难以对癌症实现实时动态检测。因此,迫切需要能够在血液、尿液或脓液等简单易得的体液样本中对肿瘤生物标志物进行无创检测。2018年,Dong等[50]利用dd PCR鉴定了包含7个KRAS突变的参考材料,发现通过dd PCR检测K R A S突变等位基因的结果与重力稀释法(gravimetrical dilution)一致,准确性优于二代测序(next generation sequencing,NGS),且未检测到任何探针的交叉反应性。此外,利用已建立的方法实现了对KRAS突变检测试剂盒的验证。Luo和Li[51]建立了一种基于cf DNA的dd PCR检测KRAS的方法,并应用于对结直肠癌患者血浆中cf DNA的检测,其检测限可达到cf DNA拷贝数的个位数,可检测到低至0.01%~0.04%的突变丰度。上述应用可以表明,d P C R在早期筛查方面具有灵敏和精确定量的优势,未来有望成为各种临床情况下疾病分析与诊断的有力工具。
2.4 、甲基化DNA的检测
有研究表明,启动子高甲基化与一些关键肿瘤抑制基因的沉默有关,并且这种甲基化通常发生在癌变的早期阶段,所以甲基化DNA检测被认为是癌症检测和诊断的重要手段[52,53]。目前,甲基化DNA的检测方法已有大量文献报道,其中基于限制性核酸内切酶的一类方法通常与PCR技术联合使用,而d PCR作为最新的PCR技术在甲基化DNA的检测中也得到了广泛应用。基于内切酶HpaⅡ对特定的DNA片段具有选择性作用的特性,Wu等[54]利用mcd PCR结合限制性核酸内切酶对甲基化DNA进行了定量检测。内切酶HpaⅡ对甲基化DNA不产生作用,而没有甲基化的DNA片段则被酶裂解,因此在进行PCR时只有甲基化DNA被检测。Van Wesenbeeck等[55]分别采用d PCR与q PCR检测福尔马林固定石蜡包埋(FFPE)样本的DNA甲基化,结果显示在高DNA和低DNA输入样品中,d PCR对甲基化DNA检测的准确性均更高,且亚硫酸氢盐不完全转化对甲基化检测准确性没有影响。
2.5、 转基因作物及食品掺假成分的检测
近年来,转基因食品饱受争议,不同的国家对转基因食品有着不同的限量标准,瑞士、芬兰等国家对转基因食品采取零容忍的态度,禁止在食品中添加任何转基因成分,所以准确灵敏的检测方法对转基因食品的监控尤为重要[56]。GB/T 19495系列标准将q PCR作为检测转基因作物的金标准,但是q PCR在检测转基因成分基质复杂的作物时会造成偏差,使检测的结果不准确,而且还容易对低浓度的转基因样品造成漏检[57,58]。同时,使用q PCR技术进行相对定量时,每种转基因成分都需要制备相应的校准品和标准曲线,这给检测带来了一定的局限。另外,对于一些深加工的转基因食品,因其在加工过程中经历了一定的物理、化学处理或者生物反应,可能会导致DNA降解或难以提取,这给检测带来了极大的挑战[59]。而d PCR作为第三代PCR技术,由于其在低丰度检测和强耐受抑制剂方面的优势,应用d PCR对转基因作物进行检测也受到了广泛的关注。Wang等[60]构建了一种双dd PCR策略,对烘焙、油炸或微波等不同加工方式处理过的米饼中的TT51-1组分进行了检测,发现定量检测效能良好。Deng等[61]利用dd PCR方法对水稻的SPS、RBE4和ppi-PPF等三个基因进行特异性定量,发现LOQ在10~20 copies/reaction之间,即使转基因成分含量低至0.1%也可以进行精确定量,其准确性和灵敏度相对于使用相同引物和探针的q PCR效果更优。Cottenet等[62]和Demeke等[63]运用d PCR技术实现了对植物转基因成分的检测和定量,且发现该方法对低丰度核酸的检测表现出很高的重复性。Dalmira等[64]使用两种荧光探针结合d PCR实现了对玉米两种转基因成分T-nos/hmg的同时检测,检测限和定量限分别是0.034%和0.08%。
随着市场监管部门对掺伪现象的不断重视,不法商家的掺伪手段也不断升级,变得更加隐蔽,给人们带来了经济上的损失,甚至需要承担食物过敏的风险。传统的感官鉴定也无法满足市场监管的需求,基于蛋白质鉴定的免疫胶体金技术和酶联免疫吸附技术在掺伪成分和产品物种亲缘性较近的情况下,容易出现交叉反应,导致检测准确性下降[65]。另外,蛋白质热处理的变性降解给蛋白质的鉴定带来了一定的难度。以检测核酸DNA为目标的PCR及其衍生技术,因简单快速、易于操作、灵敏度高和特异性好,已经被广泛用于食品中掺假成分的检测。d PCR以其精确和灵敏的特性能够对食品中掺入的微量物质进行分析鉴定,弥补了传统方法易受复杂食品基质干扰和不能精确定量的缺陷。有研究利用d PCR对羊肉中的鸡肉成分进行了检测,结果发现:相比于q PCR,d PCR对含有5%~80%鸡肉比例的羊肉的检测偏差小于9%,且鸡肉成分对羊肉的量化结果没有影响,在不同的热处理和超高压处理下均表现出较好的重复性和稳定性[34]。另外,杨华等[66]选择特异性良好的引物和探针,建立了多重d PCR检测方法对牛肉、鸡肉、鸭肉和猪肉等四种肉类成分进行鉴别判断,所测样品与已知信息完全相符,大大缩短了鉴别时间。
3 、展望
从1971年首次体外扩增DNA到1999年第三代PCR技术d PCR的发明,PCR技术得到了迅速全面的发展,并广泛应用于生物学领域的检测。d PCR作为新一代核酸定量技术,可实现对微量靶基因的绝对定量,且不受基质的干扰。但是,d PCR作为一种新兴的检测手段,还是存在着一些固有局限与不足。(1)易受污染。高灵敏度作为d PCR一大优势的同时,使得d PCR容易受外源污染造成假阳性结果,因此该技术对操作人员和实验室有着极为严苛的要求。微液滴的吸取和转移等PCR扩增后的开放式操作会导致微液滴内容物的挥发和逸出,从而使PCR产物产生气溶胶污染。比如,在dd PCR样品分散过程中,液滴的破裂会造成扩增产物的释放。因此,为保证结果的可靠性,芯片的防污染设计就显得尤为重要。Song等[67]通过构造一种由粘接在玻璃罩上的四层聚二甲基硅氧烷(PDMS)组成的纳升自吸间隔化(SPC)微流控芯片,实现了负压环境中样本的自吸,此外玻璃罩的使用也可防止样品溶液的蒸发。(2)有效动态范围小。d PCR绝对定量结果的精度受限于被检样品的浓度。当数万个检测单元达到饱和时,被检样品的浓度越高,其检测精度越低,使得d PCR检测的有效动态范围变窄。对于动态范围的扩展,最直接经济的方法是对样品进行稀释,当样品浓度高于动态范围时可使其进入支持的检测区域,但当浓度在检测下限附近时将使其超出支持的动态范围,因此需要对稀释步骤进行优化以满足对所测样本浓度的连续能力[68]。分区数量的增加也可有效扩展动态范围,如Rain Dance Technologies公司(现已被美国Bio-Rad公司收购)所生产的Rain Drop?数字PCR系统可将反应体系无限稀释至1 000万份,有效动态范围达到了6 logs。另外,Liao等[69]开发了一种新方法,通过使用能调节PCR混合物折射率的甜菜碱生产出光学透明的PCR乳剂,接着用快速3D光片荧光显微镜完成乳液的原位且高通量读数,使动态检测范围提高到与q PCR相当的数量级。(3)高成本。由于目前d PCR仪主要被几大公司所垄断,d PCR的推广和应用面临着高额的费用,尤其是配套的试剂耗材,相比于q PCR高出几十倍,这些都限制了d PCR的应用。然而,d PCR的高精度在一些应用领域的实际意义尚不清楚。在一项关于巨细胞病毒载量接近治疗阈值的移植患者纵向样本的回顾性分析中,没有证据表明d PCR比q PCR更具临床意义,会更及时地给临床医生提供正确用药的指导依据[70]。在q PCR能够达到检测要求的情况下,检测人员无论是从检测成本、时间、操作复杂程度还是使用熟悉程度等方面,都更倾向于使用技术比较成熟的q PCR。(4)不确定度。目前许多基于液滴或者芯片的商业化d PCR系统分区体积与制造商理论上规定的体积存在差异。在dd PCR系统中一般存在5%的体积不确定度,这是由液滴生成仪分区的多分散性造成的,而微流控系统则是在制造公差上存在3%的体积不确定度[71]。体积的均匀性会对精度和动态范围存在影响,特别是对于高浓度的样品[72]。(5)分区错误。样本分散时会带来不可避免的分区错误。每个反应微室或液滴理论上期待的是最多存在一个DNA模板,然而实际操作中却通常含有2个及以上的核酸模板。虽然泊松分布可以对误差进行一定程度的优化调整,但却不能消除,这会导致基因拷贝数的低估[73]。此外,在荧光监测过程中,会出现荧光检测系统无法正确区分阳性液滴和阴性液滴的情况,这可能是由扩增效率降低引起的中等荧光水平造成的[74]。阳性与阴性分区之间的荧光信号对比度偏低,也会导致液滴的错误分区。Zhu等[75]通过构建一种“准共聚焦”式激光诱导荧光(LIF)细胞术检测光路最大程度地激发液滴内的荧光信号,从而提高了阳性分区和阴性分区之间的荧光对比度。(6)通量低。作为d PCR仪的一个重要性能指标,荧光通道的多少直接决定了对同一样本中的不同靶标进行多重检测的能力,这对检测时间和检测成本造成了一定程度的影响。目前商业化的d PCR系统大部分为2~4通道,然而荧光通道的增加绝非易事,在其对应的光谱区域中存在合适染料的前提下,还必须确保与其他通道没有光谱重叠[76]。虽然单色多路复用的策略已经被用来提高检测通量,但是其受限于DNA的质量和PCR抑制剂,且在2D直方图荧光分析过程中簇的分辨率不是很高,难以从其他簇中区分终点信号[50]。
目前,d PCR系统的样品分散、热扩增和信号检测三个步骤一般是由对应功能的3台仪器分别完成,存在样本丢失和分散或转移过程污染的风险。因此,未来d PCR将打造一个高度集成化的d PCR检测平台,只由一台仪器来完成整个检测过程。在保证系统精确度的同时,d PCR将朝着高通量和自动化的方向发展,以低成本实现同时对多个目标序列的绝对定量。虽然d PCR目前存在着一些缺陷,但是随着d PCR检测技术的完善和商业化仪器的不断开发,d PCR的准确度和灵敏度也将越来越高,在未来生物学检测中的应用也将越来越广泛。
参考文献
[1] Mullis KB. Target amplification for DNA analysis by the polymerase chain reaction. Ann Biol Clin, 1990, 48:579-82
[2] Simonetti S, Chen X, Dimauro S, et al. Accumulation of deletions in human mitochondrial DNA during normal aging:analysis by quantitative PCR. Biochim Biophys Acta, 1992,
1180:113-22
[3] Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci USA, 1999, 96:9236-41
[4] Kwon HR, Kim JG, Lim HS, et al. Construction of a cDNA library of Aphis gossypii glover for use in RNAi. Entomol Res, 2018, 48:384-9
[5] Tschiedel E, Goralski A, Steinmann J, et al. Multiplex PCR of bronchoalveolar lavage fluid in children enhances the rate of pathogen detection. BMC Pulm Med, 2019, 19:132
[6] Lakatos B, HornyakA, Demeter Z, et al. Detection of a putative novel adenovirus by PCR amplfication,sequencing and phylogenetic characterisation of two gene fragments from formalin-fixed paraffin-embedded tissues of a cat diagnosed with disseminated adenovirus disease .Acta Vet Hung, 2017, 65:574-84
[7] Hussain H, Chong NFM. Combined overlap extension PCR method for improved site directed mutagenesis. BioMed Res Int, 2016, 2016:8041532
[8] Lehmann P, Ehrenstein B, Hartung W, et al. PCR analysis is superior to histology for diagnosis of Whipple's disease mimicking seronegative rheumatic diseases. Scand J Rheumatol, 2017, 46:138-42
[9] Shahdadnejad N, Mohammadabadi MR, Shamsadini M.Typing of Clostridium perringens isolated from broiler chickens using multiplex PCR. Genet 3rd Milennium,2016, 14:43
68-74
[10] Coffey J, Choudhry M, Shlossman M, et al. Multiplex real-time PCR detection and relative quantification of periodontal pathogens. Clin Exp Dent Res, 2016, 2:185-92
[11] Jiang X, Xiang J, Wang R, et al. A development strategy to fast establish the Taqman qPCR based method to detect SNP mutations. Hum Cell, 2020, 33:1331-3
[12] Emaus MN, Anderson JL. Allelic discrimination between circulating tumor DNA fragments enabled by a multiplexqPCR assay containing DNA-enriched magnetic ionic liquids.
Anal Chim Acta, 2020, 1124:184-93
[13] Fischer BM, Neumann D, Piberger AL, et al. Use of highthroughput RT-qPCR to assess modulations of gene expression profiles related to genomic stability and interactions
by cadmium. Arch Toxicol, 2016, 90:2745-61
[14] Chillin MC, Jimenez C, Garcia-Sanz R, et al. Quantitative PCR:an alternative approach to detect common copy number alterations in multiple myeloma. Ann Hematol,2017,
96: 1699-705
[15] Yang X, Ding T, Huang H, et al. Development and validation of a simple and rapid method for hepatitis C virus genotyping based on one-step RT-qPCR. Exp Ther Med, 2020,
20:2284-90
[16]赵丽青.方佩佩,唐静,等数字PCR定量检测食品中单核细胞增生李斯特氏菌方法的研究.食品安全质量检测学报, 2017, 8:4133-8
[17] Abachin E, Convers s, Falque S, et al. Comparison of reverse-transcriptase qPCR and droplet digital PCR for the quantification of dengue virus nucleic acid. Biologicals,2018, 52:49-54
[18] Koepfi C, Nguitragool W, Hofmann NE, et al. Sensitive and accurate quantification of human malaria parasites using droplet digital PCR(ddPCR). Sci Rep, 2016, 6:39183
[19] Furuki H, Yamada T, Takahashi G, et al. Evaluation of liquid biopsies for detection of emerging mutated genes in metastatic colorectal cancer. Eur J Surg Oncol, 2018, 44:975-82
[20] Tanaka T, Kobunai T, Yamamoto Y, et al. Increased copy number variation of mtDNA in an array-based digital PCR assay predicts ulcerative coliti-associated colorectal cancer. In Vivo, 2017, 31:713-8
[21] Aigrain L, Gu Y, Quail MA. Quantitation of next generation sequencing library preparation protocol eficiencies using droplet digital PCR assays--a systematic comparison ofDNA library preparation kits for llumnina sequencing. BMC Genomics, 2016, 17:458
[22] Beaver JA, Jelovac D, Balukrishna S, et al. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin Cancer Res, 2014, 20:2643-50
[23] Stevanato P, Biscarini F. Digital PCR as new approach to SNP genotyping in sugar beet. Sugar Tech, 2016, 18:429-32
[24] Manoj P. Droplet digital PCR technology promises new applications and research areas. Mitochondrial DNAA DNA Mapp Seq Anal, 2016, 27:742-6
[25] Koppel R, Bucher T. Rapid establishment of droplet digital PCR for quantitative GMO analysis. Eur Food Res Technol, 2015, 241:427-39
[26] Conte D, Verri C, Borzi C, et al. Novel method to detect microRNAs using chip-based QuantStudio 3D digital PCR. BMC Genomic, 2015, 16.1-11
[27] Rowlands V, Rutkowski AJ, Meuser E, et al. Optimisation of robust singleplex and multiplex droplet digital PCR assays for high confidence mutation detection in circulating tumour DNA. Sci Rep, 2019, 9:1-13
[28] Schuler F, Schwemmer F, Trotter M, et al. Centrifugal step emulsification applied for absolute quantification of nucleic acids by digital droplet RPA. Lab Chip, 2015, 15:2759-6
[29]叶庆张标,杨军,等.用于液体活检的新技术一数字PCR检测技术 临床检验杂志,2019, 37:564-7
[30] CaoL, Cui X, Hu J, et al. Advances in digital polymerase chain reaction(dPCR)and its emerging biomedical applications. Biosens Bioelectron, 2017, 90:459-74
[31] Sreejith KR, Ooi CH, Jin J, et al. Digital polymerase chain reaction technology-recent advances and future perspectives. Lab Chip, 2018, 18:3717-32
[32] Doi H, Takahara T, Minamoto T, et al. Droplet digital polymerase chain reaction(PCR)outperforms real-time PCR in the detection of envirnmental DNA from an invasive fish species. Environ Sci Technol, 2015, 49:5601-8
[33] Cui X, Cao L, Huang Y, et al. In vitro diagnosis of DNA methylation biomarkers with digital PCR in breast tumors .Analyst, 2018, 143:3011-20
[34] Ren J, Deng T, Huang W, et al. A digital PCR method for identifying and quantifying adulteration of meat species in raw and processed food. PLoS One, 2017, 12:e0173567

自1985年KaryMullis发明PCR技术以来,PCR技术一直是生物医学领域中重要的实验方法。随着分子生物学技术的发展,目前核酸定量的主要方法是实时荧光定量PCR(QuantitativePCR,qPCR)。数字PCR(DigitalPCR,dPCR)是近年来发展起来的新技术,是对传统P...

相对于此前的核酸定量方法,dPCR达到了前所未有的灵敏度、特异性和精确性,尤其在复杂基质及痕量样品检测方面具有独特优势,其为分子生物学、医学、微生物和环境科学等领域的研究提供了全新的技术手段和思路。...