生物化学论文


胰蛋白酶( trypsin,EC 3. 4. 21. 4) 是丝氨酸蛋白酶家族的成员,是许多生物技术领域的工具酶,如动物细胞培养过程,一些蛋白酶原活化过程等。在胰岛素的生物合成过程中,通常是以胰岛素原的形式表达,然后在体外通过羧肽酶 B 与胰蛋白酶的特异性切割,转化为起作用的胰岛素,但是目前所用到的胰蛋白酶大多数是从动物( 如猪、牛、羊等)的胰腺中直接提取,在此过程中,有可能引入 供体动物携带的致病微生物,所以急需重组胰蛋白酶的合成。在人体中,主要存在两种胰蛋白酶原,即人胰蛋白酶原-1( trypsinogen-1,记为 PRSS1) 与人胰蛋白酶原-2( trypsinogen-2,记为 PRSS2) ,二者约占人体内胰液总蛋白的 19%,由于有研究报导 PRSS2 与人体的某些病理过程密切相关,所以本研究选择PRSS1 作为研究对象,PRSS1 相对分子质量约为 24kDa,含有 247 个氨基酸,其中有 10 个 Cys。在其 N端 1 ~15 位氨基酸为胰蛋白酶原表达信号肽,16 ~23 位的 8 个氨基酸残基( APFDDDDK) 是肠激酶的识别位点,也即胰蛋白酶原活化肽( Trypsinogen acti-vation peptide,TAP) ,在体内可被肠激酶切割成为具有活性的胰蛋白酶。Kiraly 等曾尝试在大肠杆菌中直接表达胰蛋白酶原,未发现有目的蛋白分泌,通过用 Met 代替信号肽后,有以包涵体形式出现的蛋白,Chen 等在研究胰蛋白酶原活化肽的功能时,也用 Met 代替信号肽,可见,信号肽对于胰蛋白酶在大肠杆菌中的表达并非必须。若直接表达胰蛋白酶,由于其能够特异性地在 Lys 与 Arg 的 C-端切割多肽,会对宿主造成很大伤害,也大大降低了目的蛋白的表达水平。本研究借鉴前人融合蛋白表达的优势,将胰蛋白酶作为融合蛋白表达。
二硫键形成蛋白 A ( Disulfide bond formationprotein A,DsbA) 最初是由 Bardwell 等于 1991 年发现,存在于大肠杆菌周质胞腔内,主要负责催化新生蛋白质二硫键形成。DsbA 在胞内与目标蛋白融合表达,有效地提高了目的蛋白的可溶性。
当然,不是所有的 DsbA 融合蛋白均能够得到可溶性的目的蛋白。人源胰蛋白酶-1 富含二硫键,且在载体 pET-39b( + ) ( Novagen) 上含有催化二硫键形成的周质蛋白酶 DsbA 的基因,我们将人源 Tryp-sin-1 的基因片段连接到 DsbA 的 C-端,利用 DsbA与 Trypsin-1 融合表达,预期促进 Trypsin-1 蛋白中二硫键的正确折叠,实现目的蛋白的可溶性表达,并利用载体 pET-39b( + ) 所携带的凝血酶的酶切位点来实现 Trypsin-1 的活化。
1 材料和方法
1. 1 材料
大肠杆菌 DH5α 与 BL21( DE3) 感受态及 EasyPfu DNA polymerase 购自北京全式金生物技术有限公司,载体 pET-39b( + ) 购自 Novagen,Trypsin-1 全基因为北京奥科鼎盛生物科技公司合成。各种普通限制性内切酶为大连 Takara 有限公司产品; Fast-digest 内切酶与预染标准核酸相对分子质量蛋白为Fermaentas 公司产品; 质粒小量快速抽提试剂盒购自北京原平皓生物技术有限公司; 琼脂糖凝胶纯化回收试剂盒为北京博迈德科技发展有限公司产品;预染标准核酸相对分子质量为康为世纪公司; 其余试剂为国产或进口分析纯。
1. 2 实验方法
1. 2. 1 Trypsin-1 全基因密码子优化和二级结构分析
不同宿主对同一氨基酸有不同的密码子偏好性,同时 mRNA 编码区稳定性、外源蛋白的毒性对外源蛋白在原核中表达也有重要的影响。按照 Codon usage 网站提供的 E. coli 密码子使用频率表,对人源 trypsin-1( GenBank Accession No. NM_002769. 4) 的碱基进行优化设计,对密码子使用频率低的氨基酸进行同义突变。有文献报导 mR-NA 二级结构的稳定性对翻译起始效率起着至关重要的作用,进而影响蛋白的翻译,我们用 RNAstruc-ture 软件分析序列对应的 mRNA 的二级结构。
1. 2. 2 Trypsin-1 基因的扩增、克隆以及原核表达载体的构建
以含 Trypsin-1 基因的重组质粒 pGH-trypain-1为模板,设计引物进行 PCR 扩增,正向引物序列为5-aaaAGTACTATCGTTGGGGGCTATAACTG 反 向 引物序列为: 5-ccgCTCGAGTTATTAGCTATTGG CAG-CA。在正向引物和反向引物的两端分别引入 ScaI和 XhoI 酶切位点。PCR 产物经纯化后用 ScaI 和XhoI 30 ℃ 酶切 2 h,与同样经 ScaI 和 XhoI 酶切的pET-39b( + ) 质粒连接,连接产物转化大肠杆菌DH5α 后采用菌落 PCR 法和双酶切验证法筛选阳性转化子克隆,并送测序公司进行测序确定。测序正确的阳性克隆转化子质粒 pET39b-trypsin-1,转入感受态大肠杆菌 BL21( DE3) 菌种。
1. 2. 3 重组 Trypsin-1 的诱导表达与 SDS-PAGE分析
将 pET39b-trypsin-1/BL21( DE3) 重组菌接入含50 μg / mL 卡那霉素( Kan+) 的 LB 培养基中于37 ℃培养活化过夜,次日按 V( 活化菌液) ∶ V( LB 培养基) =1∶ 100 转接至新鲜的 LB 培养基中,37 ℃培养至 OD600 为 0. 6 左右,加入诱导剂 IPTG,24 ℃进行诱导,220 r/min 振荡培养 8 h。
取 100 mL 菌液,以 8000 r/min,在 4 ℃条件下离心 10 min 收集菌体,用溶液( 50 mmol/L Tris-HCl100 mmol / L NaCl pH = 8. 0) 洗涤菌体 3 次后,离心并重悬菌体,17% 的功率超声裂解 20 min。分别将裂解上清液、沉淀通过 SDS-PAGE 电泳分析,其中分离胶浓度为 15%,浓缩胶浓度为 5%,电泳后以考马斯亮蓝 R-250 染色,凝胶薄层扫描,分析目的蛋白表达情况。并尝试不同的发酵温度、诱导剂量、诱导时机以及装液量,确定最适宜培养条件。
1. 2. 4 Trypsin-1 包涵体的复性
离心收集到的菌体,按 1 L 发酵液所得菌体用20 mL 50 mmol / L Tris-HCl 重悬,8 000 r / min,4 ℃ 离心 5 min,洗涤 3 次,然后用 20 mL 50 mmol/L Tris-HCl 重悬混匀,冰上放置 30 min,17% 的功率超声裂解 20 min,4 ℃,12 000 r/min 离心 30 min,弃掉上清,用包涵体洗涤液( 3 mol/L 尿素,0. 6% Triton-X100,50 mmol / L Tris-HCl,4 mmol / L EDTA) 重悬沉淀,洗涤 30 min; 12 000 r/min 离心 30 min,弃掉上清,用包涵体洗涤液再洗 1 次,离心弃上清; 沉淀用5 mL 变性液( 50 mmol / L Tris-HCl,10 mol / L 尿素,40 mmol / L DTT,1 mmol / L EDTA) 充分溶解 3 h,4 ℃ ,12 000 r / min 离心 30 min,回收上清; 将复性液( 50 mmol/L Tris-HCl,150 mmol/L NaCl,10 mmol/LGSSG,5 mmol / L CaCl2) 缓慢加入到变性的蛋白溶液中,至尿素终浓度为 2 mol/L,4 ℃复性 12 h。复性完毕后,以50 mmol/LTris-HCl,5 mmol/L CaCl2缓冲液 4 ℃透析。
1. 2. 5 Trypsin-1 活性测定
Trypsin 的活性定义为以 N-苯甲酰基-L-精氨酸乙酯 盐 酸 盐 ( BAEE ) 为 底 物,用 pH 值 7. 8,0. 050 mol / L Tris-HCl 缓冲液配置 1. 0 mmol / L 底物溶液,光程 1 cm,反应温度 25 ℃,波长 253 nm 的条件下,OD 值递增 0. 001 min- 1,为 1 个 BAEE 单位。
酶活力单位( BAEE 单位) = ΔOD/t × 1000。
并以市售猪胰蛋白酶( 北京索莱宝科技有限公司) 为对照: 2. 5 g 猪胰蛋白酶和 0. 2 gEDTA 溶于1 L PBS 中,不含钙和镁,含酚红,过滤除菌,- 20 ℃保存,活性 250BAEE 单位 mg- 1。
取 200 μL 复性蛋白液与 20 μL 凝血酶切割缓冲液( 10 × Buffer: 200 mmol/L Tris-HCl,pH = 8. 4,1. 5 mol / L NaCl,25 mmol / L CaCl2) 混合,加入 1 μL凝血酶,室温酶切后取 200 μL 酶切溶液测定其活性。
2 结果
2. 1 Trypsin-1 基因表达质粒的构建
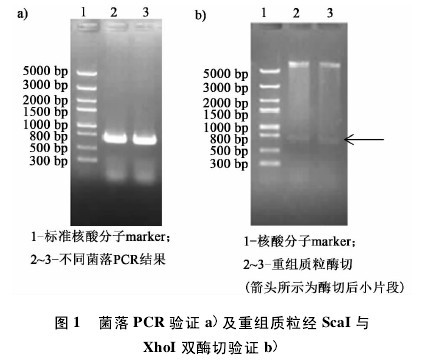
构建的重组表达质粒 pET39b-Trypsin-1,经菌落PCR[图 1a) ]和双酶切[图 1b) ]验证,得到 678 bp左右的特异性扩增条带,经测序验证,结果显示与所要构建的目的基因片段完全一致。【图1】
2. 2 Trypsin-1 在大肠杆菌 BL21(DE3)中的表达
pET39b-Trypsin-1 / BL21( DE3 ) 经 IPTG 诱导的菌液裂解物与未诱导的同样菌液裂解物在 15% 的SDS-PAGE 电泳图上对照分析( 图 2 ) ,可以看到小于 55 kDa 处有 1 条明显的表达蛋白带,但是目的蛋白主要以包涵体的形式出现在沉淀中,分子质量与理论计算的相符。优化后的摇瓶发酵条件为37 ℃,220 r / min,500 mL 摇瓶中装液量为 100 mL,IPTG诱导 10 h 包涵体量达到最大,融合蛋白约占菌体总蛋白的 40%。IPTG 的浓度对蛋白表达影响较小,试验中用量为 0. 5 mmol/L。
2. 3 Trypsin-1 的亲和纯化
重组融合蛋白 DsbA 与 Trypsin1 之间含有 6XHis·Tag,其与金属 Ni 离子具有亲和作用,以 Ni-NTA 交联的琼脂糖作为层析介质,对超声裂解离心后的上清液柱上纯化,多次试验发现洗脱峰中没有目的蛋白,其中也曾尝试用更低的温度 ( 20 和16 ℃ ) 诱导表达也未发现有目的蛋白洗脱下来。说明对于人源的胰蛋白酶,单独的与 DsbA 融合表达,不能实现融合蛋白可溶性表达的目的。改用对包涵体变性和复性的方法得到目的蛋白。
2. 4 Trypsin-1 的活化和胰蛋白酶活力的测定
用紫外分光光度计测量 ΔOD253来检测融合蛋白复性后经凝血酶活化获得的胰蛋白酶活性,15%SDS-PAGE 结果分析见图 3。酶切 12 h 后,复性的融合蛋白基本被切开,活性达到最大,为 4 个 BAEE活力单位。对于市售猪胰蛋白酶,相当于 1 L 发酵液获得有 1. 6 mg 有活性的目的蛋白。
2. 5 重组基因表达的 mRNA 二级结构软件分析结果
用 RNAstructure 软件分析了经过密码子优化后重组基因对应的 mRNA 的二级结构,图 4 优化后在翻译起始部位的碱基配对比优化前明显减少了,这样有利于翻译效率的提高。
3 讨论
不同来源的 Trypsin 基因片段在不同的宿主菌中已成功构建,其中在 Pichia Pastoris 中实现了猪源、牛源Trypsin 的可溶性表达,但是产量都并不高。其中在 E. coli 表达人源胰蛋白酶源-1,由于其富含二硫键,目前所获得产物主要以包涵体的形式出现,通过体外重折叠所获得的有活性的胰蛋白酶,得率相当低。但是 E. coli 由于发酵周期短、遗传背景清楚、成本低廉等优势,更被倾向优先作为宿主菌。本实验用所构载体 pET39b-tryp-sin-1 转化 BL21 ( DE3 ) 经 IPTG 诱导表达及发酵条件优化后,虽然没有实现预期的可溶性表达,但是在包涵体中实现了 DsbA-Trypsin 的大量表达。
所得沉淀超声破菌后经缓冲液的洗涤、变性、复性,用凝血酶切割,得到了有活性的胰蛋白酶,说明部分胰蛋白酶形成了正确的构象。以市售的猪胰蛋白酶为参照( 2. 5 g/L,250 U/mg) ,每 1 L 发酵液获得了约 1. 6 mg 有活性的 Trypsin,复性过程操作简便,适合放大过程。包涵体的复性与复性起始蛋白浓度、溶液的 pH 值、缓冲液的组分等条件有关,从图 3 中可以看出本实验在包涵体变性方面损失了大量的蛋白,在复性方面有待做进一步的探索。
本试验通过包涵体表达获得了大量的胰蛋白酶,由于胰蛋白酶有较多的二硫键,通过包涵体的洗涤、变性和复性,最后得到的有活性的蛋白得率较低。和包涵体蛋白复性相比,纯化可溶表达蛋白相对更节约成本和时间,利于放大生产,目的蛋白得率也较稳定和较高。本实验室利用载体 pET-39b( + ) 在 E. coli BL21( DE3) 中已成功表达了可溶性的牛肠激酶轻链 EKL片段,但是通过本试验证明同样的手段并不适用于人源胰蛋白酶的表达。本试验为后续在 E. coli 中探讨人源胰蛋白酶的可溶性表达可以提供一定的借鉴,如另有文献报道 Ds-bA 的突变型与真核蛋白 IGF-I,3C 蛋白酶,TGFβ-2和 GFP 等蛋白融合表达,有效的提高了目的蛋白的可溶性,值得尝试。此外,本试验表达的胰蛋白酶主要用于工业生产胰岛素原的激活反应,所以没有进行进一步的纯化试验,而直接用于胰岛素原的切割,这样可以减少多步纯化工序,节约成本。
真菌、细菌对抗生素的耐药性引起了全世界范围的普遍担忧, 导致巨大的财力、物力花费在寻找新型抗生素上, 但效果甚微。一些抗生素对普通的感染作用效果差, 引起大众对不可治愈的恐慌。...