���\������

����ժ Ҫ������ƽ�ĸSaccharomyces cerevisiae�Ǵ�л����������Ҫ������֮һ���Ƚ��Ļ���༭�����Ѿ����㷺Ӧ������ƽ�ĸϸ����������ƺ��������Ż���༭�����ķ��ٷ�չ�����ڻ�������ø��ͬԴ����Ļ���༭���������ͻ���༭ϵͳ����������ж���ƽ�ĸ����༭������ԭ����Ӧ�ý������ܽᣬ�����������ƽ�ĸ����༭���������ں�������ø��MegNs��ZFNs��TALENs�Ȼ�����༭ϵͳ�������ܺ������˻���CRISPR/Casϵͳ����Դ��л;��������Ϻͻ������ģ����༭�������о���չ��������ƽ�ĸ����༭������Ӧ��ǰ���ͷ�չ���������չ����
�����ؼ��ʣ�����ƽ�ĸ; ����༭; CRISPR; �������;
����Abstract����Saccharomyces cerevisiae is one of the most important hosts in metabolic engineering. Advanced gene editing technology has been widely used in the design and construction of S. cerevisiae cell factories. With the rapid development of gene editing technology, early gene editing technologies based on recombinase and homologous recombination have been gradually replaced by new editing systems. In this review, the principle and application of gene editing technology in S. cerevisiae are summarized. Here, we first briefly describe the classical gene editing techniques of S. cerevisiae. Then elaborate the genome editing system of MegNs, ZFNs and TALENs based on endonuclease. The latest research progress is especially introduced and discussed, including the CRISPR/Cas system, multi-copy integration of heterologous metabolic pathways, and genome-scale gene editing. Finally, we envisage the application prospects and development directions of Saccharomyces cerevisiae gene editing technology.
����Keyword����Saccharomyces cerevisiae; gene editing; CRISPR; multi-copy integration;
����Ϊʵ��������Դ����ϸ��ѧƷ����Ȼ����ȵ���ɫ���ɳ������죬���ô�л������ϳ�����ѧ��������ϸ��������һ����ǰ;�IJ���[1]����Ϊһ�ֵ��͵��������ϵͳ����ƽ�ĸSaccharomyces cerevisiae����������³���ԡ��Կ��̷��������������ԡ����̲����ĸ�Ч�Ժ��ϵİ�ȫ��(Generally recognized as safe,GRAS)����ѧ���о���ҵ�������Ѿ��õ��˹㷺Ӧ��[2,3,4]��������ƽ�ĸ��Ҫ���ڷ�������Ʒ(���������ͷ������)�;ƾ�����(��ơ�ơ���)�ȵ���������һ���Ա��ܿ�����ȼ���Ҵ��͵�ϸ�����ķ������������Ŵ�л���̼����ķ�չ����ƽ�ĸ��Խ��Խ������������л��ᡢ�����ᡢ�����ᡢҩ�õ��ס���ҵø�Ƽ��Ͷ����֬����(PUFAs)[5]�ȣ���Щ��Ʒ���㷺Ӧ����ʳƷ��ҽҩ�����Ϻͻ�����ҵ[6]�����⣬��ƽ�ĸ����Ȼ�����������Դ�ϳɷ���Ҳ��ʾ���˶��ص����ơ���ԭ��������ȣ���ƽ�ĸ���н�Ϊ�����ķ��������ϵͳ��ϸ����ϵͳ(���������塢��������ø�塢���������߶������Һ��)��Ϊ��Ȼ���������ϳ��ṩ�˲�ͬ�Ļ����ͼ��[7,8]���ر�������Դ���︴�Ӵ�л;���Լ�����Ĥ��ϵ���ϸ��ɫ��P450����øʱ���ֳ���Խ������[9,10,11,12,13]��Ŀǰ������ֲ�������л�����Ѿ�����ƽ�ĸ��ʵ�ֺϳɣ��������[11]�����㰱����[14]������[15]�ͻ�ͪ��[16]�ȡ���ɹ��������ǿ�űҩ��������ǰ��������������ƽ�ĸ�еĺϳɣ������ߴ�25 g/L������ʵ�ֹ�ҵ������[17]��

����“���-����-����-ѧϰ”�Ǵ�л���̵�һ����̺�ʵ��ϸ����������Ԥ��Ŀ��ıؾ�֮·[18](ͼ1)�����У���������֤��Ƶı�Ҫ�ֶΡ�Ȼ��������Խ��Խ��Ļ���������ƽ�ĸ��ʵ�ֺϳɣ�ϸ�������Ĺ�����;���Ż�����Ҳ��¶����ȱ�ݺ;��ޣ�������Ҫ�ڵ�һλ����ж�β���[14]�����ѳ�Ϊ��л���̵����ٲ��衣��ˣ�������Ч�Ļ���༭�������ڱ��С�����༭�����Ƕ�Ŀ�������λ��������������Σ����������ó������롢�滻������ĵ�ͻ��[19]������༭�������ִ����\������������Ҫ�����ã���Ҫ��Ի���ͻ�����Ԥ��λ�ý��о�ȷ��Ч�ı༭������[20,21]����ͳ�Ļ���༭�����Ǹ���DNAͬԴ����(Homologous recombination,HR)ԭ����������Դ�������Ƭ���ó�������Ŀ�����Ƭ�Σ������ַ���Ч�ʽϵͣ�������������ĸ�����10–9–10–6[22]����ƽ�ĸ�г��õĻ���༭ϵͳ��Cre/Lox Pϵͳ��ͬ���ǻ���ͬԴ����ԭ���Ļ���༭����[23]��Ϊ������������Ч�ʣ�������˫�������Ѿ���֤�����������ͬԴ���鷢���ĸ���[24,25]�����ڴˣ����ͻ���༭�����Ѿ����������ã���Ҫ����4�֣��鳲��������ø(Meganucleases,Meg Ns)ϵͳ��пָ����ø(Zinc finger nucleases,ZFNs)ϵͳ��ת¼����������ЧӦ�����ø(Transcription activator-like effector nucleases,TALENs)ϵͳ��CRISPR (Clustered regularly interspaced short palindromic repeats)��ص�ϵͳ(��1)[19]����Щ�¼����Ŀ�����Ӧ�ã�����شٽ��˴�л������ϳ�����ѧ�ķ�չ�����Ķ���ƽ�ĸ����༭�����ķ�չ����״�������ܽᣬ�������ڻ���༭���������ͱ༭������ͨ���༭�����ȡ�������ƽ�ĸ����༭�����ķ�չǰ��������չ����
����ͼ1 ��ƽ�ĸϸ����������
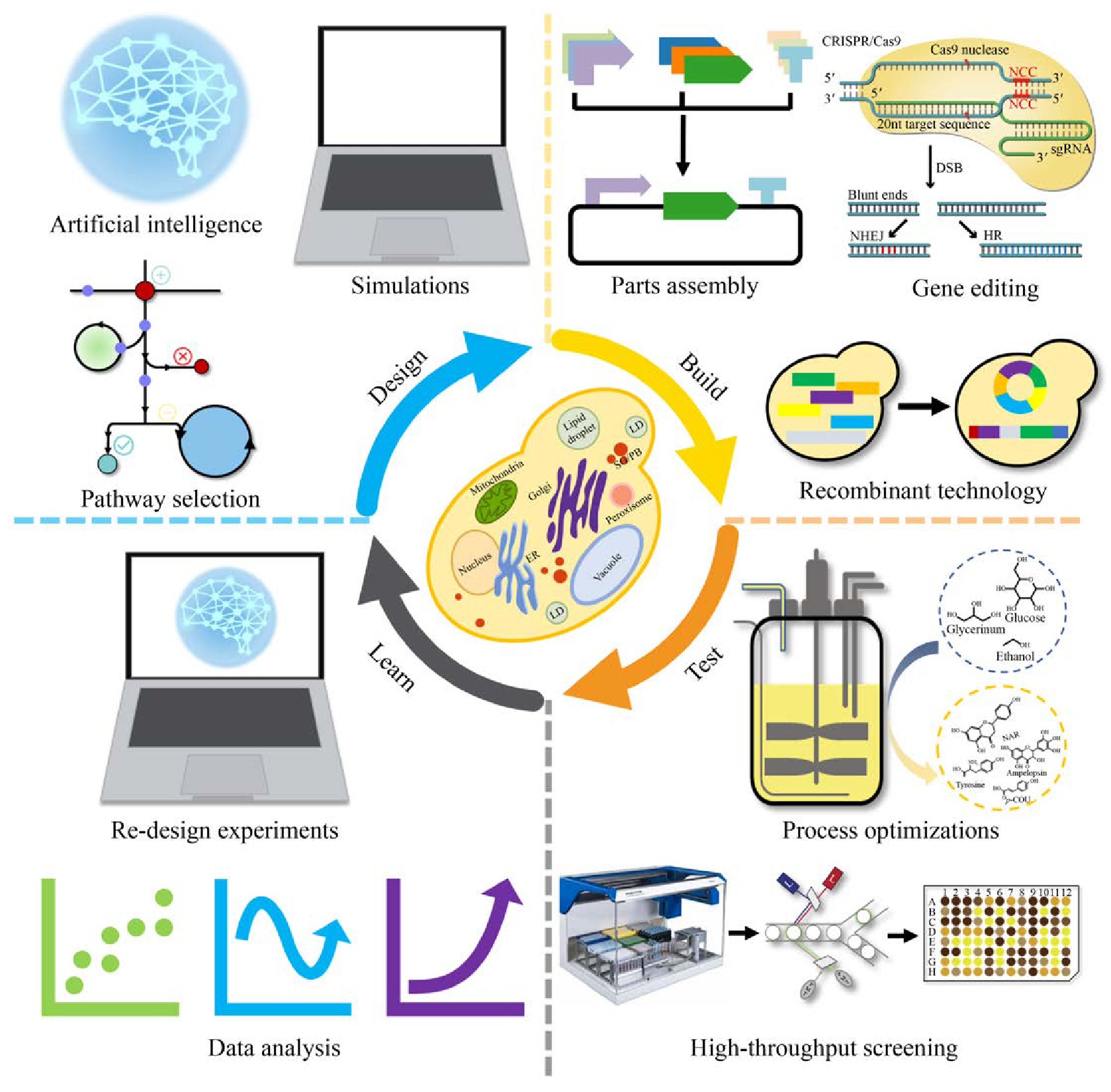
����Fig.1 Construction of S.cerevisiae cell factory.
������1 ����༭��������ƽ�ĸ�е�Ӧ��
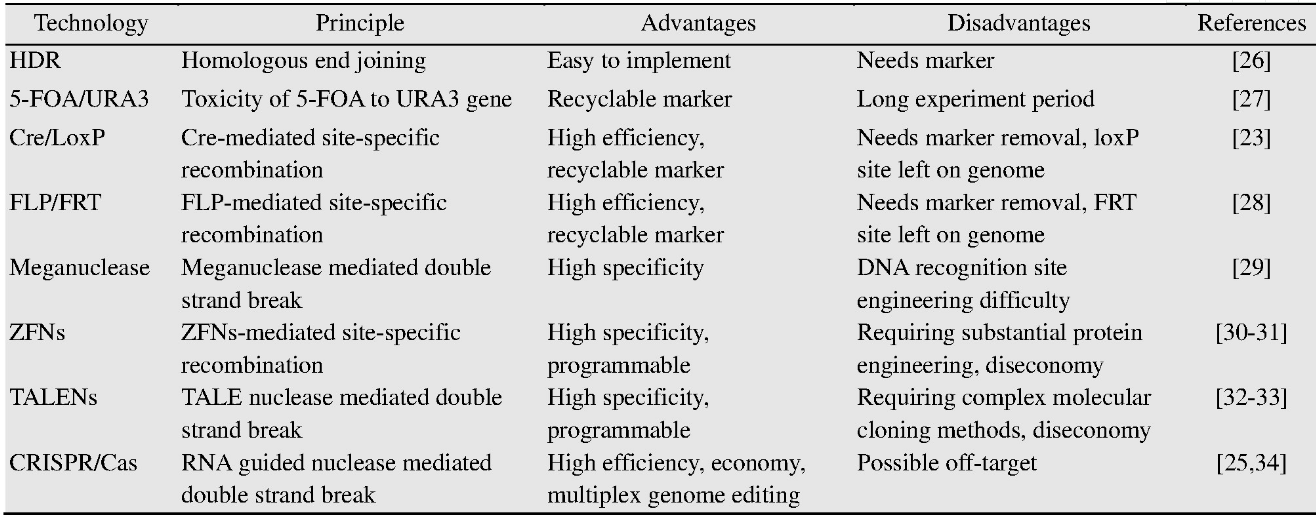
����1 ����ƽ�ĸ�������༭����
����20����70��������̵ķ�չΪ������༭��������;��[35]��������ƽ�ĸ����༭������Ҫ����ͬԴ�����ɸѡ��ǡ����У�ͬԴ���鼼������δ���˵�ͬԴDNAƬ����Ϊģ�壬��ʵ��Ŀ���������ӡ��滻��ʧ��[36]�����־���Ļ�����༭��������ϸ�����������еõ��˹㷺Ӧ�á�����ƽ�ĸ����Ҫ�������ֻ��Ʋ���DNA˫����������ͬԴ����(Homology-directed repair,HDR)(ͼ2A)�ͷ�ͬԴĩ������(Nonhomologous end-joining,NHEJ)(ͼ2B)[24]��HDR�����ô���Ŀ�����ͬԴ���е�DNA��Ŀ��λ��ʵ�־�ȷ�Ļ���༭����NHEJ����ϸ��������һЩ��Ԫ��������������������̣��������ɻ��������IJ����ȱʧ�����»����Ķ���ͻ��[37]��
���������ʵ��ѡ���ǩѭ�����õļ���������������5-FOA/URA3��ɸѡ�����ͻ�������ø�ĸ�ɸѡϵͳ�����У�5-FOA/URA3��ɸѡ��������5-��������(5-fluoroorotic acid,5-FOA)��URA3������ƽ�ĸ���̾�����ж��Ե��ص㣬��ɸѡURA3������ƽ�ĸ���̾���(ͼ2C)[38]����������ø���õ�ѡ���ǩ����ϵͳͬ���Ѿ����㷺Ӧ�õ�����ϸ�����̵Ĺ�����[39]��������Դ����ƽ�ĸ2μ����������������ϵͳFLP/FRTϵͳ[28]��Դ���ɾ���P1��Cre/lox Pϵͳ[40](ͼ2D)����5-FOA/URA3��ɸѡ������ȣ�������ϵͳ���и��ߵı༭Ч�ʡ��������ַ��������������ظ�����(Inverted repeats,IRs)����������֮����ڻ�����������һ���ظ����еĸ���(lox P��FRTλ��)���ڶ���ظ�����ѡ���ǩ���»�����IJ��ȶ�������[41]��Ϊ�˱��������ظ����жԺ�������༭��Ӱ�죬��ͻ���FRT��lox Pλ��ɱ����������[42]����������ϵͳ�ܴ�̶���������ɸѡ��ǩ�����Ե����ƣ�������ȱ���ڵ�һת��������ͬʱ�������������εĿɿ�������ʹ��ɸѡ��ǩ����л�����Ȼ��һ����ʱ�Ĺ���[43]�����⣬�����ڹ��̾����б�����Ӧ������ø��������ҪЯ����Ӧ����ø������������ж���ת����������ʵ������������Ȼ��Щ�����ij��������ϸ������������Ч�ʣ�����ʹ��ɸѡ��ǩ�Ի�������������ģ����ҶԻ�������ж�Ŀ��༭ʱ�������롣
����ͼ2 �������༭����
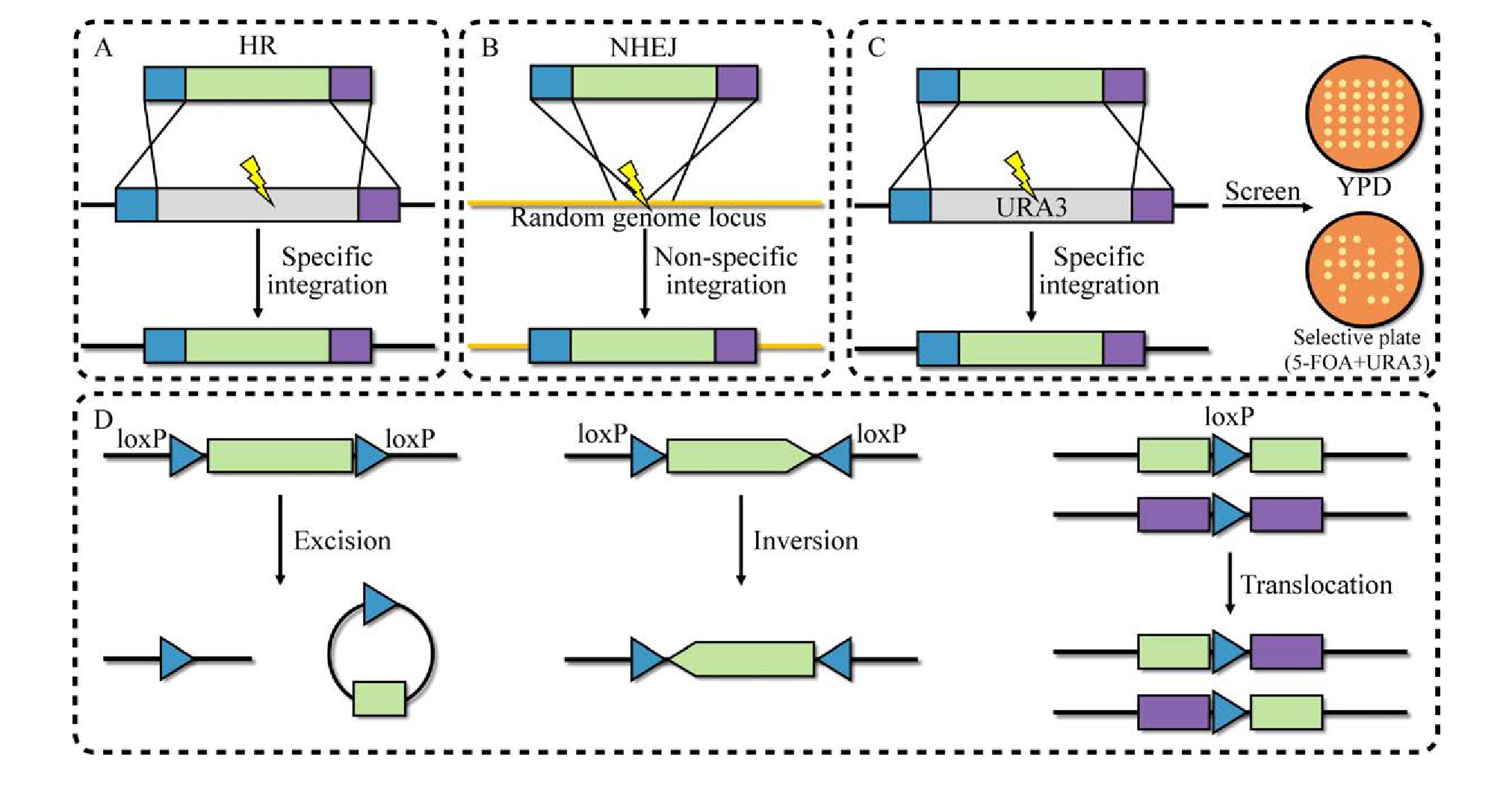
����Fig.2 Classic gene editing technology.(A) Homologous recombination connection.(B) Non-homologous terminal connection.(C) 5-FOA/URA3 negative screening technology.(D) Cre/lox P technology.
����2�� ����Meg Ns��ZFNs��TALENs�Ļ�����༭ϵͳ
�������Ż���༭������Ѹ�ͷ�չ�����ڻ���༭ϵͳ�Ѿ������ͻ���༭ϵͳ��������ں�������ø�Ļ���༭���������鳲��������ø(Meg Ns)������пָ������ø(ZFNs)����[44]��ת¼����������ЧӦ�����ø(TALENs)[45]������CRISPR��ؼ���[46]�ȡ�ͨ����Ŀ������������˫������(Double strand break,DSB)������������ж��������༭������DSB�ڽ�ĸ�о��������ԣ������Щ���������Ͽ�������ɸѡ��ǩ���Σ�����Ŀ��λ����о�ȷ���졣
����2.1�� Meg Ns����
����Meg Ns�Ǿ��нϴ�ʶ��λ���������������Ǻ�������ø�����ʶ��14������Ե�˫��DNA���У�����ƽ�ĸ�еĢ�-Sce��鳲��������ø����ʶ��18 bp������[47]��Ϊ��ʵ�ֶ�Ŀ�����ı༭��ͨ����Ҫ���鳲��������øʶ��λ��������������Ⱦɫ��Ŀ��λ�ã����Meg Ns���ڻ�����Ŀ��λ�㸽������DSB�����������༭Ч�ʡ�Kuijpers�����â�-Sce����Ŀ��Ⱦɫ��λ������˫�����ѣ�ʹ����Ƭ�ε�����Ч����ߵ�95%[41]��
����2.2�� ZFNs����
����ZFNs������ָ��пָ����ø�鵼�Ļ���༭������пָ����ø�ǽ������ڻ�����༭���˹��ϳ�����������ø����Ҫ����������ɰ����������Ժ�������øFok��ķ�������DNA�и�ṹ��Ϳ�ʶ��������DNA��пָ����(Zinc finger protein,ZFP)(ͼ3)[48]��ZFP�ɴ�����Cys2-His2пָ����ģ�鹹�ɣ�ͨ������Cys2-His2пָ�ɴ�Լ30����������ɣ�Ϊһ��α-������2�������βƽ�й���β-β-α�ṹ[48]���ʵ�����пָ�ṹ���������Ը�ȷ��ʶ��Ŀ�����У��������пָ���Ի�����Ŀ��Ƭ�ε�������ʶ��[49]��ÿ��пָ����ģ�����ѡ����ʶ��DNA���е�3�������(Base pair,bp)����ͨ����α-�����л���DNA��(Major groove of DNA)����ã��γɼ�������ԽӴ�[50]��Fok����Ҫ�γɶ�����ſ�����������е�Ŀ�������Խ�����Ч�Ļ���༭����ˣ�ͨ����Ҫ������ZFNs������DNA���ʺϵķ����ϲ���λ��Ŀ�����У���ʹ������ʶ��ļ������������һ��[50]��
����ͼ3 ���ں�������ø�Ļ���༭����
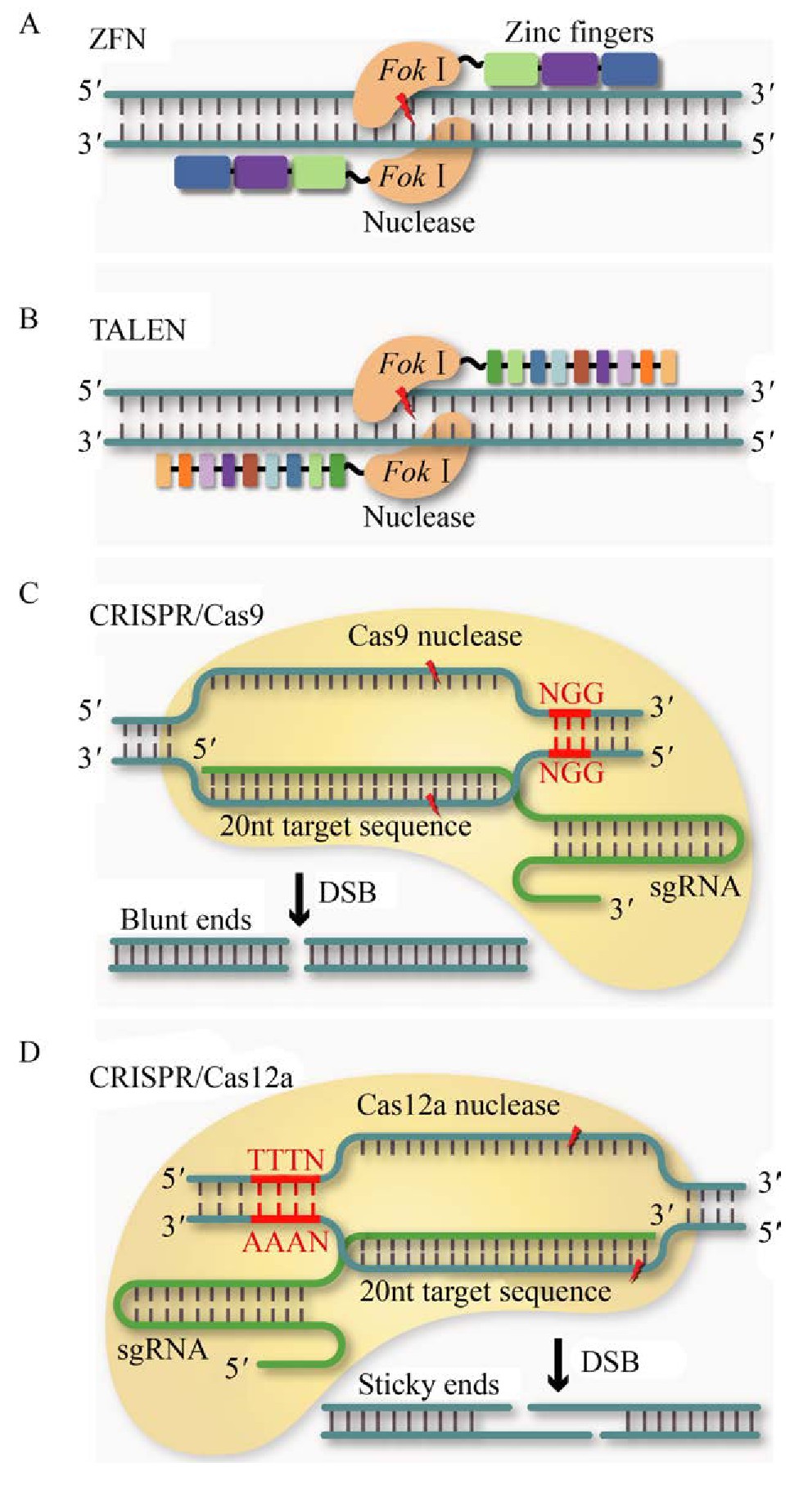
����Fig.3 Gene editing technology based on endonuclease.(A) ZFN system (B) TALEN system (C) CRISPR/Cas9system (D) CRISPR/Cas12a system.
������ƽ�ĸ��Ϊ�����ģʽ����Ѿ���������֤�����ZFNs��Ч��[47]����ɥʧ���ܵ�MEL1����������ϵ���ĸ�������У��������Я����ѡZFNs���������Ա༭Ч��[30]�����л��Ե�ZFNs��Ŀ��DNA���˫�����ѣ����ͨ��ͬԴ����ķ�ʽ���������[30]��
����2.3�� TALENs����
����TALENsͬ����һ�ֹ��̻�����ø������һ���������Ե�DNA�и����һ��DNA�����Խ����(ͼ3)����ZFNs�������ƣ�Ҳ��Ҫ���������Ժ�������øFok��ķ�������DNA�и�ṹ��Ի�������б༭[51]��DNA�����Խ������ת¼����������ЧӦ����(TALE)�߶ȱ��ص��ظ�������ɣ������ֲ��Ƶ�����Xanthomonas campestris�б�����[52]��TALE���ظ��ṹ��ͨ����34��������л���ɣ����е�12���͵�13���ظ����пɱ�˫������л�(Repeat-variable di-residues,RVDs)������DNA��ϵ�������[53]��ÿ��RVD����ʶ��һ���ض��ĺ����ᣬ�Ӷ��γ�һ���̶���DNAʶ�����(NIΪ�����ʣ�HDΪ����ण�NGΪ������ण�NH��NNΪ������)�������˳����װ�Խ���κλ�����Ŀ������[54]����ͬ��RVD��Fok���������γ�TALENs����ʶ���Ŀ�����г���ͨ��Ϊ14–20 bp,2��Fok������γɶ���������Ի�������б༭����ZFNs��ȣ�TALENs�Ĺ���������Լ�����Ӧ�÷�Χ���㣬����Ժ�Ч��Ҳ����[55]����������ɱ���Ը߰�����������һ�ֵͳɱ������ܹ����Ļ�ȡ���������ٲ���������TALENs������ƽ�ĸ�У�����ZFNs��TALENs�����Ի�����Ŀ��λ�����˫�����Ѻ�ͨ����ԴHR��NHEJ������ʵ�ֶ�Ŀ�����λ��Ķ���༭��Ȼ�����Բ�ͬ�����������ʱ��������ƺ����ºϳ�ZFNs��TALENs���ס�
����3 ������CRISPR�Ļ�����༭����
����CRISPR/Cas����༭������2013����ֵ���С����RNA�鵼��һ�ְ��������༭���¼������ѳ�Ϊ����ZFNs��TALENs�����յ��������༭��һ��DZ�ڼ�����Ч�ķ���[56]��Ŀǰ��CRISPR/Casϵͳ�Ѿ���Ϊ�����������еĻ�����༭���ߣ���Ӧ���ڸ����������У�����ϸ���������ֲ��Ͳ��鶯��[24]��CRISPR/Casϵͳ��һ����Ӧ������ϵͳ��Դ��ϸ���ž�����������������������߷�������[57]��CRISPR�������Ѿ��ڴ�Լ40%��ϸ����90%�Ĺž������з���[58]��CRISPR/Casϵͳ�ɶ��Cas����IJ����Ӻ�һ��DZ���CRISPR-RNAs (cr RNAs)��ɡ�cr RNA���������ظ����м���ֿ����������ظ����С��������ظ������Ǵ�����DNA�л�õ��������Ƭ�Σ��������DNA���е�������ʶ�𡣵�����DNA����ʱ���µ�����ʶ������Ҳ���Ա����ϵ�cr RNA�У��������������ٴα�ͬһ����DNA����[59,60]��
����3.1�� CRISPR������༭����ԭ��������
����CRISPR/Casϵͳ������DNA���и���Ҫ��3���Σ�������Դ����DNA����cr RNA�ϳɺ��и��߽�[57,61]�����ȣ�CRISPR/Casϵͳ��һ��Cas�����Ӽ���ػ�������ֵ�����DNA���н���ʶ�𣬲�������DNA������Ϊ�������ظ����в��뵽����������CRISPR���в��֣��Լ�����һ���µ�CRISPR���С�����ʶ��������������ε�ǰ���������ڽ�����(Protospacer adjacent motif,PAM)����ͬ��Դ��CRISPRϵͳ��PAM����Ҳ���ڲ��졣���CRISPR���б�ת¼���ӹ��ɵ�����cr RNA��ÿ��cr RNA����������ǰ���ֵ�����DNA�������Ӧ��RNAƬ���Լ�����CRISPR�ظ����С����cr RNA������Cas������ɵĸ��������������DNA���н����и��߽⡣
����CRISPR/Casϵͳ���ж����ԣ�������DNAΪ�е��ϵͳ����RNAΪ�е��ϵͳ����DNA��RNAΪ�е��ϵͳ[57]��������ЧӦ�����������Խ�CRISPR/Casϵͳ��Ϊ�����࣬1��(������͢���)��ʹ�ö൰�����2��(���͡����ͺ͢���)���õ�������ø(��Cas9��Cas12��Cas13)��1���2��CRISPR/Casϵͳ�������ṹ�Ϸֱ�������Ƶ��ص㡣���͡����ͺ͢�������4–7��Cas�����ǻ���ɶ൰��ЧӦ��������͡����ͺ͢������ɵ�����ṹ����ɡ������������ŶԸ�ϵͳ�ṹ���ܵIJ�������̽���о���CRISPRϵͳ�ѳ�Ϊһ�ֳ����Ч�Ļ���༭���ߣ�����Ӧ����Ϊ�㷺����CRISPR/Cas9ϵͳ�����˵�CRISPR/Cas12a(Cpf1)ϵͳ��
����3.2�� CRISPR/Cas9ϵͳ
��������Դ����ŧ�����Streptococcus pyogenes�Ģ���CRISPR/Cas9ϵͳΪ�������ǶԿ���������DNA�ķ���ϵͳ[39]����ϵͳ����Cas9���ס�cr RNA��tracr RNA (trans-activating cr RNA)���ɵĸ�����[47](ͼ3)��Cas9���װ�����������ø�ṹ��HNH��Ruv C�����Ƿֱ��и���cr RNA��20��������(nt)�������л����ͷǻ�����DNA�����γ�ƽĩ��DNA˫������[62]�����˹������cr RNA��tracr RNA����ϳ�Ϊһ��g RNA(guide RNA)���ӡ�ͨ�����������ã����۵��ɲ���˫����RNA�ṹ����Cas9��Ϸ��ӹ��ܡ���ˣ�����Ҫͨ���ı�g RNA��Ŀ�������DNA�����Խ�����б����ʵ�ֻ���༭��Ŀ�ġ�����Ŀ�����е���һ��Ҫ����ͬ����ҪPAM������ʹCas9����ʶ���ϡ�Cas9�����������ٽ�PAMλ�������Ŀ�����н��м��в���DSB�����ͨ����ƽ�ĸ�е�ͬԴ�����ƣ������ʵ�ֻ�����Ŀ������ȱʧ�����ϡ�
����3.3�� CRISPR/Cas9ϵͳ����ƽ�ĸ�е�Ӧ��
����CRISPR/Cas9ϵͳ�Ѿ���Ϊ��ƽ�ĸ������༭�ĸ����ԺͶ�ܲ��ԡ�Ŀǰ��CRISPRϵͳ�ѱ�����Ϊ���л����ó������ϡ�ת¼���ź�ת¼�������������л�����еĴ���������������������CRISPR/Cas9ϵͳʵ��[63]��CRISPR/Cas9ϵͳͨ�����̻��ĵ�������RNA (Single guide RNA,sg RNA)����Cas9����øЧӦ���γɸ�����Ӷ��и�PAM��������3 bp���Ļ����飬���DSB[64]��������DNA�����������DNAͬԴ����ʱ����ƽ�ĸ���ͨ��DSBʵ�ֶԻ�����������Ի���༭��
����Di Carlo���״α���������CRISPR/Casϵͳ����ƽ�ĸ��������ó�[65]����û��ɸѡ��ǩ������£�ͨ��90 bpͬԴ����ʵ����CAN1������ó���Jako?i��nas�ȿ�����������ƽ�ĸ�����������ó���CRISPR/Cas9ϵͳ��������ͬʱ��5����ͬ�Ļ�����λ������ó�[66]��ͨ����ת��g RNA��������Ӧ������ͬԴ����Ƭ�Σ�����������5������ı༭Ч�ʴﵽ50%–100%�����̾���������������Ұ���;��������41����Eau Claire�ȳɹ���17������β-���ܲ��غϳɵĻ���Ƭ��һ�������ϵ���ƽ�ĸ�������У�����DNAƬ�ν���50 bp��������һ��֤����CRISPR/Cas9ϵͳ��ͬԴ�������ϵ���Ч��[67]��Zhang�Ƚ�t RNA��������g RNA����������g RNA-t RNA-array CRISPR/Cas9 (GTR-CRISPR)ϵͳ��ͬʱ�ó�8�������Ч�ʴﵽ87%�����������˻���༭Ч��[20]��
�������ڹ�ҵ��ƽ�ĸ���ԣ�����������ܸ��Ӹ��ӡ�ͬʱ����������ʳƷ����ҵ�������칤ҵ����ƽ�ĸ����ʳƷ��ȫ�ĽǶȳ�����������ƽ�ĸ��ӦЯ���п��Ա�ǩ�Ͷ��Գɷֱ��������˶����Ŵ�������Ҫ�����Ͻ�����ͨ���ϸ�İ�ȫ�����ۺɻ������С�Ȼ���������λ����Ĵ��ں�ȱ��ɸѡ��ǼӴ����Ŵ��������Ѷ�[68]����ˣ�CRISPR/Cas9ϵͳ���ޱ�DZ༭��ʽ���ʺ�Ӧ�õ�������Ͷ�幤ҵ����Ļ��������[69]��Lee������CRISPR/Cas9ϵͳ�����˹�ҵ��ĸ����JHS200��Ӫ��ȱ����ͻ���壬������Ӧ������������Silver grassˮ��Һ���������Ҵ����о���[68]��ͨ������ľ��;����NADH����ø�������ά���Ҵ��������������Ҵ������ﵽ55.5 g/L[68]��Zhang������CRISPR/Cas9ϵͳ�ڹ�ҵ�����ƽ�ĸATCC 4124��ʵ����Ӫ����ǩURA3��TRP1��LEU2��HIS3���ó�[70]��Lian��ͨ�����ӱ���g RNA�����Ŀ���������˻����ó�Ч�ʣ��ó����������������������4�������Ч��Ϊ100%[71]����Щ�о�������CRISPR/Cas9ϵͳ�ڶ�幤ҵ���Ӧ���ǿ��еġ�
����CRISPR/Cas9ϵͳ�������Ǿ��л���༭�Ĺ��ܣ������̻���CRISPR/Cas9ϵͳ������ʵ���������ع��ܣ�������������ƺͼ���[63]����Cas9����ø�ṹ��HNH (H840A)��Ruv C(D10A)����ͻ�䣬�ɵ��������ṹ��ʧ��Ӷ�ʹ����øɥʧ�и����[72,73,74]�������ͻ������һ��λ�㣬����ʹCas9���׳�Ϊ�п�ø����ֻ�и�˫��DNA�е�һ����������һ��g RNA����ʹ��ʱ���ɷֱ�������Ŀ��DNA���ϲ����пڡ�
����3.4�� CRISPR/Cas12a(Cpf1)ϵͳ
����CRISPR/Cas12a(Cpf1)��һ�����ͻ���༭���������������ڵ����ЧӦ�����Ż����2�����CRISPRϵͳ[75](ͼ3)������Դ�ڰ��������Acidaminococcus spp.BV3L6��As Cpf1����Դ��ë�ݿƾ�Lachnospiraceae bacterium ND2006��Lb Cpf1Ϊ����Cas12aʶ���PAMλ��ΪĿ��DNA 5’�˴�����T������(��5’-TTTN-3’)����g RNA��������Ŀ��DNA�ض�λ���ϲ�ִ���и��[75]����Sp Cas9��ȣ�As Cpf1��Lb Cpf1�ķ�������С����������ϸ������ֻ��Ҫcr RNA����tracr RNA���������и�����и�DNA��RNA������[76]�����⣬���з�ʽ��Cas9Ҳ�нϴ���졣Cas9����ͬһ��λ��ͬʱ����DNA���ӵ�˫��������γ�ƽĩ�ˣ���Cas12a(Cpf1)���к��γ�������ͬ���ȵ������γ����ĩ��[75]��ͬʱ��������PAM���������ʶ��Ŀ�����е�������ʹCRISPR/Cas12a(Cpf1)ϵͳ�Ѱ��ʸ���[77]��Li��ʹ��CRISPR/Cas12a(Cpf1)ϵͳ�ɹ�ɾ������ƽ�ĸ����������������֮�䳤��38 kb��Ƭ�Σ�֤��CRISPR/Cas12a(Cpf1)ϵͳ��������ƽ�ĸ�Ļ������[78]��Verwaal���о���3�ֲ�ͬ��Դ��Cas12a(Cpf1)����ƽ�ĸ������༭�Ĺ��ܣ����еİ����������Acidaminococcus spp.BV3L6�������ָ�����˿��Francisella novicida U112������Դ��Cas12a(Cpf1)�ڵ�����༭�Ͼ�����CRISPR/Cas9ϵͳ�൱�ı༭Ч�ʣ����⣬��Դ��ë�ݿƾ�Lachnospiraceae bacterium ND2006��Lb Cas12a(Cpf1)��ʵ�ָ�Ч�Ķ��ػ�����༭����ͬʱ��3������ܲ��ػ����������ϵ�3����ͬ�Ļ�����λ��[79]��
�������Ͽ��Կ��������۶���CRISPR/Cas9ϵͳ����CRISPR/Cas12a(Cpf1)ϵͳ��˵��sg RNA���������ö���������Ҫ�ġ�CRISPRϵͳ�ѹ㷺Ӧ���ڶ��ֲ�������Ļ���༭�С�Ȼ����CRISPRϵͳ�ľ�ȷ����༭һֱ���Ѱ�ЧӦ���š�sg RNA���ȶ������������������CRISPRϵͳ��������Ҫ���á���ˣ���ø�������sg RNA��CRISPR�༭�ɹ��Ĺؼ�����֮һ��
����3.5 ���Ż�sg RNA�����
����Ϊ�˽����Ѱ�ЧӦ�����ڼ����������sg RNA���������Ѿ������������CRISPRϵͳ�ľ�ȷ��[80]�����ǣ����ڲ�ͬ��ƽ�ĸ������������һ�����죬sg RNA�����������վ���������ݴ��ھ����ԣ���һ���̶���������CRISPRϵͳ�ı༭Ч�ʡ���ˣ��������ԵĻ�����������ͬ��ʮ����Ҫ�����⣬��Cas9�п�ø��˫sg RNA���ϲ��ԣ�ͬ�����Խ����Ѱ�Ч��[81]����Ҫע����ǣ�����sg RNA��Ҫ�㹻������Ҫ��λ���������ϣ��Զ�Ŀ��������˫�����ѡ����⣬����sg RNA�Ŀ�Ͷ��CRISPR��ϵͳ(Multi-functional genome-wide CRISPR,MAGIC)�Ѿ���������Ŀ�������о�ȷ�༭�͵��ػ������ˮƽ[46]��
����3.6 ������༭��
��������༭��(Base editor,BE)������ɥʧ���Ե�Cas9 (d Cas9��Cas9�п�ø)������Ѱ�ø(Cytidine deaminases)�������Ѱ�ø�ںϣ��Ի�������е����ͻ��ļ���[82]������sg RNA�İ��������������ڲ�����DNA˫�����ѵ�����¶�Ŀ��������������Եı༭��ɥʧ���Ե�Cas9������Ѱ�ø�ںϣ��ɽ����������Ѱ�Ϊ�������գ�ʹ��������ԭ�е�C·Gת��ΪT·A�������������Ѱ�ø(Deoxyadenosine deaminase)�ںϣ��ɽ����������Ѱ��γ��������գ�ʹ��������ԭ�е�A·Tת��ΪG·C�����ü���༭���İ���DNA�ձ��Ѿ�����ƽ�ĸ�еõ�֤ʵ[83]�����ǣ�����༭��Ҳ����һ���ľ����ԣ�������Ϊ���ŵ�������Ŀǰֻ�����ڲ�ͬ���֮���ת��(Transition)������ʵ�ֲ�ͬ���֮��ĵ�(Transversion)������ֻ��ʵ����श���ऺ����ʶ����ʵĸı䣬���������श����ʺ����ʶ���वĸı䡣����һ����Ǽ���༭����RNAˮƽ�Լ�ȫ������ˮƽ�ϴ��ڽϸߵ��Ѱ�ЧӦ��
����4 ����Դ��л;���������
�������ڵ�����λ�����Դ�������Ϸ�ʽ�Ѿ�Ӧ�õ���ƽ�ĸ������������[84]��Ȼ�������ڿ����������ƣ���Դ����ı���ˮƽ��Ŀ��������Ҳ���ܱ����ơ�������ƽ�ĸ�������λ������Ϸ�ʽ�ɽ�һ�������Դ����Ŀ����������ʺ�Ӧ���ڴ�л���̵ĸ�����[85]����ƽ�ĸ�г�������Դ�������ϵĶ��λ����Ҫ�к�����DNA (Ribosome DNA,r DNA)��δλ���Tyת����(ͼ4)��
����r DNA��Ԫ������ת¼��(5S��35S)��������ת¼���(NTS1��NTS2)��ɣ�һ��r DNA��Ԫ���ȴ�ԼΪ9.1 kb����Լ��150���������[86]������r DNAλ������Ϸ�ʽ���ѱ��㷺Ӧ�õ����̾���Ĺ����С�Dai��[87]���Ƚ���Դ�������Panax quinquefolius��Pg DDS��Pg PPDS�����Ͻ�Arabidopsis thaliana��At CPR1�������ϵ�����BY4742��r DNAλ��ʵ����ԭ�˲ζ����ĺϳɣ����ں���ʵ�����ȶ����Zhang��[88]��β-����֬���ϳ�;���Լ�ǿ�����﹩Ӧ;�����ϵ�r DNAλ�㣬���̾���SGibʵ����β-����֬���ĸ�ˮƽ�ϳɡ����Zhu��[15]�����ڹ��̾���SGib�Ͻ��и��죬������ϸ��ɫ��P450ø(CYP72A154��CYP88D6)��һ��ϸ��ɫ��P450��ԭøATR1�Ļ������ϵ��������ϣ��ɹ��ϳ��˸ʲݴ��ᡣPark��[89]��װ�����춡������ϳ�;����r DNAλ�㣬q PCR��������3����;���������ϣ��춡��������227.2 mg/L����δ������춡����������һ�£�������Դ;����������ȶ�������r DNAλ�㡣
����ͼ4 ��ƽ�ĸ����Դ��л;���������
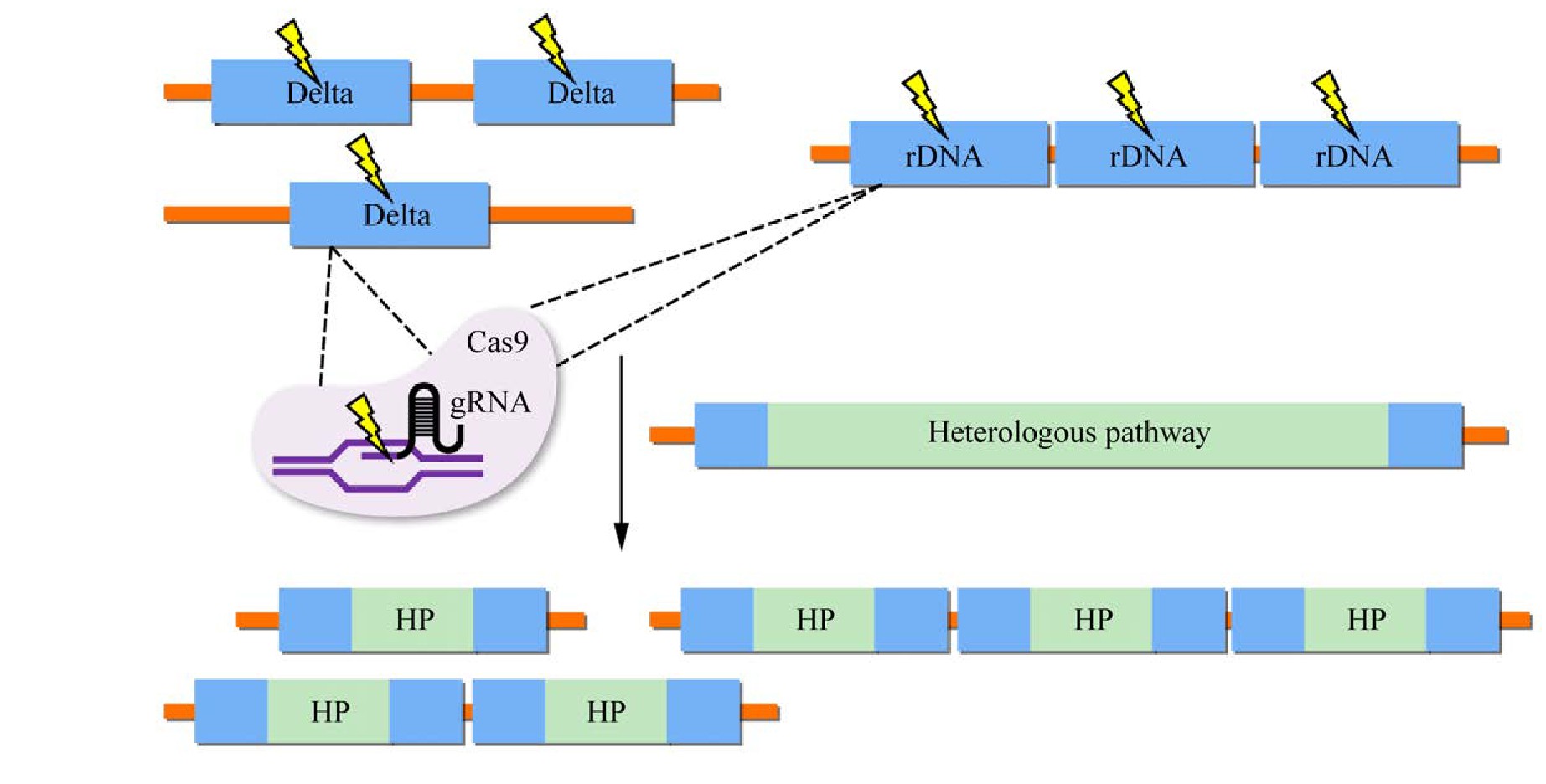
����Fig.4 Multi-copy integration of heterologous metabolic pathways in S.cerevisiae.
������ƽ�ĸ�д��ھ���ת��������Ty����(Ty elements)��ÿ�����г��Ⱦ���6 kb���ң����˷ֱ���г�Լ0.33 kb��ͬ���ظ����С�Ty���ӱ���Ϊ�����壬��Ty1��Ty5�����ڽ�ĸ�������Ͽ������϶࣬����Ϊ��Դ����IJ���λ��[60]��Shi������Di-CRISPRϵͳ��ľ������;����(R,R)-2,3-����������ϳ�;�������ϵ�Tyת���ӵ�δ�����У�ʵ���˴�����Դ����ϳ�;���ĸ�Ч�ޱ�����ϡ���ϵͳͨ���Ի�����δλ�����DSB�����������18��������24 kb DNAƬ�Σ��о������ϸߵ�;�������������ľ�ǵ������ʺ�(R,R)-2,3-�������IJ���[25]�������о�����������Դ;�������ϵ���ƽ�ĸ������߿���λ����о�DZ������Ϊ��Դ;�����ϵ춨�˻�����
����5 ���������ģ�Ļ���༭
����������ƽ�ĸ��л����ʮ�ָ��ӣ������������ı༭����������Ŀ������������������ͨ��������༭��������ʵ�ֶ������Զ���������졣���⣬��ƽ�ĸ������͵���Ⱦɫ����ƽ�ĸ�ĺϳɣ�Ϊ������������������ѧ�ṩ�˲ο���
����5.1 ����ͨ������༭����
��������Ȼ������ɸѡ�����õ��������Ŀ�껯��������ͺͶԶ��ӹ�ҵ�����������������أ�ͨ��������ֱ�����ڹ�ҵ��ģ��������ˣ���Ҫ��������л�����ˮƽ�ĸ���ͽ����������㹤ҵ������Ȼ������ͳ���췽������ͨ����������ϸ�������Ĺ����������Զ����豸�ͻ�����༭�����ķ�չ�����ָ�ͨ��ɸѡ(Highthroughput screening,HTS)�������������Ը�ͨ����ʽ�ڻ�����ˮƽ����ϸ�������������ؽ�ϸ��������Ϊ���������Ͳ�������ṩ����������������ϸ����������������[90,91,92]��
�����������������д��ģ��ͨ�����죬���Ի�þ��ͻ��⣬�پ�����ͨ��ɸѡ�����ʵ���ض�Ŀ�껯�����������Ϊ�˸������ع��������Ŀ⣬Ŀǰ�Ѿ������˻��������϶����Զ�����(Multiplex automated genome engineering,MAGE)�ĸ�ͨ������༭����[93]�������MAGE��Ӧ���ڶԴ˾�Ⱦɫ���϶������λ��Ŀ����������죬��ʵ�ִ��ģ��̺ͽ���[94]���������MAGE�Ľ�ĸ�Ѿ۽鵼�����鹤��(Yeast Oligo-Mediated Genome Engineering,YOGE)�����������ػ����鹤��(Eukaryotic multiplex genome engineering,e MAGE)Ҳ��Ӧ������ƽ�ĸ���̻�������[95,96]��e MAGE���ڲ�����DSB������¶���ƽ�ĸ�Ķ����ʵ�־�ȷ�༭���Ρ�ͨ������ƽ�ĸ�������븴�Ʋ�ĺ����������ĹѺ����ᣬʵ���˴���40%����������Ч��[96]�����ѭ��ͬʱ��������ڵĶ��λ������˹��̾���ͻ��Ч�ʣ��÷��������˽�100���ͻ���塣��β-���ܲ���;���������ӡ��������к���ֹ�ӽ��б༭�������β-���ܲ��ز�����ͬ�Ĺ��̾��ꡣ���⣬����CRISPRϵͳ��Cas EMBLR�����ɽ�ͬԴ���е�Ŀ��DNAƬ�����ϵ���������ض�λ�㣬ʵ����ƽ�ĸ���ڶ�����ޱ����װ[97]��Liu�������һ������ر�̻�������Կ��Ƹ��ӱ��͵ķ���(Multiplex navigation of global regulatory networks,MINR)����������MINR�Ŀ�[98]��ͨ���Ҵ���/��������������ʵ�飬������ͻ���岻��������Ҵ���/���������������Ҵ�������Ũ��Ҳ��Ұ���;��������2����Guo�Ȼ���CRISPR/Cas9ϵͳ������һ��ȫ�������Ŀ⣬�ڲ�ͬ�Ļ��������£���315�������ϲ��С�����Ķ������Ҫ�Խ���������[99]��
����5.2 ���˹��ϳɻ�����
����DNA�ϳɺ���װ�����ķ�չ���ٽ���Ⱦɫ����˹�������ȫ���������ţ�ʹ����������;����ϸ���������������˹��ϳɳ�Ϊ����[100]��“�˹��ϳ���ƽ�ĸ������”�ƻ�(Sc2.0)��һ��������Ŀ��ּ����ƺ���һ����ȫ�ϳɵĽ�ĸ������[101]��2011�꣬Dymond��֤���˻�ѧ�ϳ���ƽ�ĸ����Ⱦɫ���ұ�(syn��R)�͢���Ⱦɫ�����(syn��L)�Ŀ�����[102]��2014�꣬Annaluru�ȳɹ������װ�˵�һ��������ƽ�ĸȾɫ����Ⱦɫ��(syn��)��ʹ���Ⱦɫ�峤�Ⱦ���272 871 bp[100]����ĿǰΪֹ��Sc2.0���˳�Ա�Ѿ������syn��[103]��syn��[104]��syn��[105]��syn��[106]��syn��[107]Ⱦɫ��Ĺ��������⣬����Ⱦɫ����ƽ�ĸ���˹���������̽��������Դ�����ͬ��������Ҫ��ֵ[108]����ƽ�ĸ��������ƺ��ع�������ϸ������ȫ�¹��ܺͽ���DZ����Ϊ��ȫ��̽��һ���л����ṩ�˿���[101,108]��
����6�� �ܽ���չ��
������ƽ�ĸ��Ϊ��������Ѿ�ʵ���˻�ѧƷ��ֲ��μ���л�������Դ�����ʵȵĺϳɡ�������Щ����ʮ����Ҫ����Ŀǰ�ֻ�����ƽ�ĸ�IJ�Ʒ������ˮƽ��δ�ﵽ��ҵ������Ϊ��ʵ��Ŀ�����ĸ�ˮƽ����ϳɣ�ͨ����Ҫ�漰��ʮ��ø�ٷ�Ӧ����ˣ��������Դ��л;������ƽ�ĸ֮���Э������Ϊ�ؼ�����Ҫ���ǵ���Դ;���ı����л������ǰ���﹩Ӧ�ʹμ���л������ǿ�ȶ�������Ӱ������ء���Ȼ��ƽ�ĸ�Ѿ����о������������������������л�͵���������Ȼȱ���������������⡣ͨ������༭�����Ľ�һ����չ�����������ܼ���л����Ľ����ؽ�ʹ���ǶԽ�ĸ�������֪�õ���һ����������
�������ͻ���༭���ߵĿ��ٷ�չ���Ѿ�������������ƽ�ĸ�����鹤�̵��ٶȺ�Ч�ʡ��봫ͳ����༭��ȣ�����CRISPRϵͳ�ļ����ڻ���༭����Դ��л;����װ�ȷ�������ʾ�����ƣ���������ɸѡ��������ͬʱ���ж����༭������ؼ�������Դ��л;������ͻ���ͻ������ڣ�ʹ�������������л��������Ż���Ϊ���ܡ���Ȼ������CRISPRϵͳ�Ļ���༭����������һЩ���⣬�磺����Ҫ��Ŀ�������и���ʱ��������Ҫ��ƶ������������У��Ա�֤����ʵ��ȷ�Ļ���༭����ˣ���������༭��Ч�ʺ;��ȳ�Ϊ����Ҫ�������ǣ�����CRISPRϵͳ�Ľ�һ����չ������Cas���Ŀ�������ʹ�ü����ڽ���ũҵ����ҵ��ҽѧ�����о��и�������Ӧ��ǰ����
�������⣬�ڹ��̹�����������Ҫ�Ե���ϸ�����ж�θ��죬��ʹϸ����������Ŀ������Ч�ʺͲ����ﵽ���ˮƽ������������˹����ܵȼ���������ֶ�����������Ŀɿ��Ժ�Ч������˸���Ҫ�������ģ�Ļ���༭���������ѧϰ�Ľ����ںϻ��ڴ���������ʡ����ڶ�θ����Ȼ�ᵼ��ʱ�����ڱ䳤�;������ģ�����ڵ���ʵ����ͬʱ��ɴ����Ļ���༭������Ҳ������ϸ�����������ٶȵĹؼ����ء�
���������
����[1] Mougiakos I,Bosma EF,De Vos WM,et al.Next generation prokaryotic engineering:the CRISPR-cas toolkit.Trends Biotechnol,2016,34(7):575-587.
����[2] Qin JF,Zhou YJ,Krivoruchko A,et al.Modular pathway rewiring of Saccharomyces cerevisiae enables high-level production of L-ornithine.Nat Commun,2015,6:8224.
����[3] B?er E,Steinborn G,Kunze G,et al.Yeast expression platforms.Appl Microbiol Biotechnol,2007,77(3):513-523.
����[4] Lyu XM,Zhao GL,Ng KR,et al.Metabolic engineering of Saccharomyces cerevisiae for de novo production of kaempferol.J Agric Food Chem,2019,67(19):5596-5606.
����[5] Veen M,Lang C.Production of lipid compounds in the yeast Saccharomyces cerevisiae.Appl Microbiol Biotechnol,2004,63(6):635-646.
����[6] Lian JZ,Chao R,Zhao HM.Metabolic engineering of a Saccharomyces cerevisiae strain capable of simultaneously utilizing glucose and galactose to produce enantiopure (2R,3R)-butanediol.Metab Eng,2014,23:92-99.
����[7] Xu XH,Liu YF,Du GC,et al.Microbial chassis development for natural product biosynthesis.Trends Biotechnol,2020,38(7):779-796.
����[8] Paddon CJ,Keasling JD.Semi-synthetic artemisinin:a model for the use of synthetic biology in pharmaceutical development.Nat Rev Microbiol,2014,12(5):355-367.
����[9] Yang JZ,Liang JC,Shao L,et al.Green production of silybin and isosilybin by merging metabolic engineering approaches and enzymatic catalysis.Metab Eng,2020,59:44-52.
����[10] Li MJ,Kildegaard KR,Chen Y,et al.De novo production of resveratrol from glucose or ethanol by engineered Saccharomyces cerevisiae.Metab Eng,2015,32:1-11.
����[11] Galanie S,Thodey K,Trenchard IJ,et al.Complete biosynthesis of opioids in yeast.Science,2015,349(6252):1095-1100.
����[12] Siddiqui MS,Thodey K,Trenchard I,et al.Advancing secondary metabolite biosynthesis in yeast with synthetic biology tools.FEMS Yeast Res,2012,12(2):144-170.
����[13] Suastegui M,Guo WH,Feng XY,et al.Investigating strain dependency in the production of aromatic compounds in Saccharomyces cerevisiae.Biotechnol Bioeng,2016,113(12):2676-2685.
����[14] Liu QL,Yu T,Li XW,et al.Rewiring carbon metabolism in yeast for high level production of aromatic chemicals.Nat Commun,2019,10:4976.
����[15] Zhu M,Wang CX,Sun WT,et al.Boosting11-oxo-β-amyrin and glycyrrhetinic acid synthesis in Saccharomyces cerevisiae via pairing novel oxidation and reduction system from legume plants.Metab Eng,2018,45:43-50.
����[16] Lv YK,Xu S,Lyu YB,et al.Engineering enzymatic cascades for the efficient biotransformation of eugenol and taxifolin to silybin and isosilybin.Green Chem,2019,21(7):1660-1667.
����[17] Paddon CJ,Westfall PJ,Pitera DJ,et al.High-level semi-synthetic production of the potent antimalarial artemisinin.Nature,2013,496(7446):528-532.
����[18] Palazzotto E,Tong YJ,Lee SY,et al.Synthetic biology and metabolic engineering of actinomycetes for natural product discovery.Biotechnol Adv,2019,37(6):107366.
����[19] Yang ZL,Blenner M.Genome editing systems across yeast species.Curr Opin Biotechnol,2020,66:255-266.
����[20] Zhang YP,Wang J,Wang ZB,et al.A g RNA-t RNAarray for CRISPR-Cas9 based rapid multiplexed genome editing in Saccharomyces cerevisiae.Nat Commun,2019,10:1053.
����[21] Simon AJ,D’Oelsnitz S,Ellington AD.Synthetic evolution.Nat Biotechnol,2019,37(7):730-743.
����[22] Hsu PD,Lander ES,Zhang F.Development and applications of CRISPR-Cas9 for genome engineering.Cell,2014,157(6):1262-1278.
����[23] Lv XM,Wang F,Zhou PP,et al.Dual regulation of cytoplasmic and mitochondrial acetyl-Co Autilization for improved isoprene production in Saccharomyces cerevisiae.Nat Commun,2016,7:12851.
����[24] Li L,Liu XC,Wei KK,et al.Synthetic biology approaches for chromosomal integration of genes and pathways in industrial microbial systems.Biotechnol Adv,2019,37(5):730-745.
����[25] Shi SB,Liang YY,Zhang MM,et al.A highly efficient single-step,markerless strategy for multi-copy chromosomal integration of large biochemical pathways in Saccharomyces cerevisiae.Metab Eng,2016,33:19-27.
����[26] Shao ZY,Zhao H,Zhao HM.DNA assembler,an in vivo genetic method for rapid construction of biochemical pathways.Nucleic Acids Res,2009,37(2):e16.
����[27] Gao S,Lyu YB,Zeng WZ,et al.Efficient biosynthesis of (2S)-naringenin from p-coumaric acid in Saccharomyces cerevisiae.J Agric Food Chem,2020,68(4):1015-1021.
����[28] Park YN,Masison D,Eisenberg E,et al.Application of the FLP/FRT system for conditional gene deletion in yeast Saccharomyces cerevisiae.Yeast,2011,28(9):673-681.
����[29] Chevalier BS,Stoddard BL.Homing endonucleases:structural and functional insight into the catalysts of intron/intein mobility.Nucleic Acids Research,2001,29(18):3757-3774.
����[30] Doyon Y,Mc Cammon JM,Miller JC,et al.Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases.Nat Biotechnol,2008,26(6):702-708
����[31] Kim YG,Cha J,Chandrasegaran S.Hybrid restriction enzymes:zinc finger fusions to Fok��cleavage domain.Proc Natl Acad Sci USA,1996,93(3):1156-1160.
����[32] Zhang GQ,Lin YP,Qi XN,et al.TALENs-assisted multiplex editing for accelerated genome evolution to improve yeast phenotypes.ACS Synth Biol,2015,4(10):1101-1111.
����[33] Christian M,Cermak T,Doyle EL,et al.Targeting DNA double-strand breaks with TAL effector nucleases.Genetics,2010,186(2):757-761.
����[34] Vincent JJ,Martin,Leanne,et al.A highly characterized synthetic landing pad system for precise multi-copy gene integration in yeast.ACSsynth biol,2018,7(11):2675-2685.
����[35] Rothstein RJ.One-step gene disruption in yeast.Methods Enzymol,1983,101:202-211.
����[36] Schultz C,Lian JZ,Zhao HM.Metabolic engineering of Saccharomyces cerevisiae using a trifunctional CRISPR/Cas system for simultaneous gene activation,interference,and deletion.Methods Enzymol,2018,608:265-276.
����[37] Bibikova M,Carroll D,Segal DJ,et al.Stimulation of homologous recombination through targeted cleavage by chimeric nucleases.Mol Cell Biol,2001,21(1):289-297.
����[38] Ko N,Nishihama R,Pringle JR.Control of 5-FOAand 5-FU resistance by Saccharomyces cerevisiae YJL055W.Yeast,2008,25(2):155-160.
����[39] ��ʫԨ,�Թ���,����.�ϳ�����ѧ�������о���չ--DNA�ϳɡ���װ�������༭.���﹤��ѧ��,2017,33(3):343-360.Li SY,Zhao GP,Wang J.Enabling technologies in synthetic biology-DNA synthesis,assembly and editing.Chin J Biotech,2017,33(3):343-360 (in Chinese).
����[40] Gueldener U,Heinisch J,Koehler GJ,et al.Asecond set of lox P marker cassettes for Cre-mediated multiple gene knockouts in budding yeast.Nucleic Acids Res,2002,30(6):e23.
����[41] Solis-Escalante D,Van Den Broek M,Kuijpers NGA,et al.The genome sequence of the popular hexose-transport-deficient Saccharomyces cerevisiae strain EBY.VW4000 reveals Lox P/Cre-induced translocations and gene loss.Fems Yeast Res,2015,15(2):fou004.
����[42] Storici F,Lewis LK,Resnick MA.In vivo site-directed mutagenesis using oligonucleotides.Nat Biotechnol,2001,19(8):773-776.
����[43] Mans R,Van Rossum HM,Wijsman M,et al.CRISPR/Cas9:a molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae.FEMSYeast Res,2015,15(2):fov004.
����[44] Carroll D.Genome engineering with zinc-finger nucleases.Genetics,2011,188(4):773-782.
����[45] Mussolino C,Morbitzer R,Lütge F,et al.A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity.Nucleic Acids Res,2011,39(21):9283-9293.
����[46] Lian JZ,Schultz C,Cao MF,et al.Multi-functional genome-wide CRISPR system for high throughput genotype-phenotype mapping.Nat Commun,2019,10:5794.
����[47] David F,Siewers V.Advances in yeast genome engineering.FEMS Yeast Res,2015,15(1):1-14.
����[48] ��ҫ,��Ө��,������,��.����༭�����ķ�չ����ս.���﹤��ѧ��,2019,35(8):1401-1410.Liu Y,Xiong YZ,Cai ZZ,et al.Development and challenges of gene editing technology.Chin JBiotech,2019,35(8):1401-1410 (in Chinese).
����[49] �Ű�ѩ,������,���.�������μ����о���չ.���﹤��ѧ��,2015,31(8):1162-1174.Zhang BX,Sun QX,Li HF.Advances in genetic modification technologies.Chin J Biotech,2015,31(8):1162-1174 (in Chinese).
����[50] Li HY,Yang Y,Hong WQ,et al.Applications of genome editing technology in the targeted therapy of human diseases:mechanisms,advances and prospects.Signal Transduct Target Ther,2020,5:1.
����[51] Li T,Huang S,Jiang WZ,et al.TAL nucleases(TALNs):hybrid proteins composed of TALeffectors and Fok��DNA-cleavage domain.Nucleic Acids Res,2011,39(1):359-372.
����[52] Boch J,Scholze H,Schornack S,et al.Breaking the code of DNA binding specificity of TAL-Type��effectors.Science,2009,326(5959):1509-1512.
����[53] Montague TG,Cruz JM,Gagnon JA,et al.CHOPCHOP:a CRISPR/Cas9 and TALEN web tool for genome editing.Nucleic Acids Res,2014,42(W1):W401-W407.
����[54] Streubel J,Blücher C,Landgraf A,et al.TALeffector RVD specificities and efficiencies.Nat Biotechnol,2012,30(7):593-595.
����[55] Wei CX,Liu JY,Yu ZS,et al.TALEN or Cas9-rapid,efficient and specific choices for genome modifications.J Genet Genomics,2013,40(6):281-289.
����[56] Gaj T,Gersbach CA,Barbas��CF.ZFN,TALEN,and CRISPR/Cas-based methods for genome engineering.Trends Biotechnol,2013,31(7):397-405.
����[57] Zhang F.Development of CRISPR-Cas systems for genome editing and beyond.Quart Rev Biophys,2019,52:e6.
����[58] Gupta D,Bhattacharjee O,Mandal D,et al.CRISPR-Cas9 system:A new-fangled dawn in gene editing.Life Sci,2019,232:116636.
����[59] Barrangou R,Fremaux C,Deveau H,et al.CRISPRprovides acquired resistance against viruses in prokaryotes.Science,2007,315(5819):1709-1712.
����[60] Jensen NB,Strucko T,Kildegaard KR,et al.Easy Clone:method for iterative chromosomal integration of multiple genes Saccharomyces cerevisiae.FEMS Yeast Res,2014,14(2):238-248.
����[61] Shmakov S,Smargon A,Scott D,et al.Diversity and evolution of class 2 CRISPR-Cas systems.Nat Rev Microbiol,2017,15(3):169-182.
����[62] Nishimasu H,Ran FA,Hsu PD,et al.Crystal structure of Cas9 in complex with guide RNA and target DNA.Cell,2014,156(5):935-949.
����[63] Lian JZ,Hamedi Rad M,Hu SM,et al.Combinatorial metabolic engineering using an orthogonal tri-functional CRISPR system.Nat Commun,2017,8:1688.
����[64] Gasiunas G,Barrangou R,Horvath P,et al.Cas9-cr RNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria.Proc Natl Acad Sci USA,2012,109(39):E2579-2586.
����[65] Di Carlo JE,Norville JE,Mali P,et al.Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems.Nucleic Acids Res,2013,41(7):4336-4343.
����[66] Jako?i��nas T,Bonde I,Herrg?rd M,et al.Multiplex metabolic pathway engineering using CRISPR/Cas9in Saccharomyces cerevisiae.Metab Eng,2015,28:213-222.
����[67] Eau Claire SF,Zhang JZ,Rivera CG,et al.Combinatorial metabolic pathway assembly in the yeast genome with RNA-guided Cas9.J Ind Microbiol Biotechnol,2016,43(7):1001-1015.
����[68] Lee YG,Jin YS,Cha YL,et al.Bioethanol production from cellulosic hydrolysates by engineered industrial Saccharomyces cerevisiae.Bioresour Technol,2017,228:355-361.
����[69] Stovicek V,Borodina I,Forster J.CRISPR/Cas system enables fast and simple genome editing of industrial Saccharomyces cerevisiae strains.Metab Eng Commun,2015,2:13-22.
����[70] Zhang GC,Kong II,Kim H,et al.Construction of a quadruple auxotrophic mutant of an industrial polyploid Saccharomyces cerevisiae strain by using RNA-guided Cas9 nuclease.Appl Environ Microbiol,2014,80(24):7694-7701.
����[71] Lian JZ,Bao HZ,Hu SM,et al.Engineered CRISPR/Cas9 system for multiplex genome engineering of polyploid industrial yeast strains.Biotechnol Bioeng,2018,115(6):1630-1635.
����[72] Gilbert LA,Horlbeck MA,Adamson B,et al.Genome-scale CRISPR-mediated control of gene repression and activation.Cell,2014,159(3):647-661.
����[73] Jinek M,Chylinski K,Fonfara I,et al.Aprogrammable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity.Science,2012,337(6096):816-821.
����[74] Ran FA,Hsu PD,Lin CY,et al.Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity.Cell,2013,154(6):1380-1389.
����[75] Zetsche B,Gootenberg JS,Abudayyeh OO,et al.Cpf1 is a single RNA-guided endonuclease of a Class 2 CRISPR-Cas System.Cell,2015,163(3):759-771.
����[76] Fonfara I,Richter H,Bratovi?M,et al.The CRISPR-associated DNA-cleaving enzyme Cpf1also processes precursor CRISPR RNA.Nature,2016,532(7600):517-521.
����[77] �,����.��һ��������༭ϵͳCRISPR/Cpf1.���﹤��ѧ��,2017,33(3):361-371.Yang F,Li Y.The new generation tool for CRISPRgenome editing:CRISPR/Cpf1.Chin J Biotech,2017,33(3):361-371 (in Chinese).
����[78] Li ZH,Liu M,Lyu XM,et al.CRISPR/Cpf1facilitated large fragment deletion in Saccharomyces cerevisiae.J Basic Microbiol,2018,58(12):1100-1104.
����[79] Verwaal R,Buiting-Wiessenhaan N,Dalhuijsen S,et al.CRISPR/Cpf1 enables fast and simple genome editing of Saccharomyces cerevisiae.Yeast,2018,35(2):201-211.
����[80] ���,л����.ֲ��CRISPR������༭�������½�չ.���﹤��ѧ��,2017,33(10):1700-1711.Li H,Xie KB.Recent progresses in CRISPRgenome editing in plants.Chin J Biotech,2017,33(10):1700-1711 (in Chinese).
����[81] Cho SW,Kim S,Kim Y,et al.Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases.Genome Res,2014,24(1):132-141.
����[82] Komor AC,Badran AH,Liu DR.Editing the genome without double-stranded DNA breaks.ACSChem Biol,2018,13(2):383-388.
����[83] Nishida K,Arazoe T,Yachie N,et al.Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems.Science,2016,353(6305):aaf8729.
����[84] Lyu XM,Ng KR,Lee JL,et al.Enhancement of naringenin biosynthesis from tyrosine by metabolic engineering of Saccharomyces cerevisiae.J Agric Food Chem,2017,65(31):6638-6646.
����[85] Lian JZ,Mishra S,Zhao HM.Recent advances in metabolic engineering of Saccharomyces cerevisiae:new tools and their applications.Metab Eng,2018,50:85-108.
����[86] Venema J,Tollervey D.Ribosome synthesis in Saccharomyces cerevisiae.Annu Rev Genet,1999,33:261-311.
����[87] Dai ZB,Liu Y,Zhang XN,et al.Metabolic engineering of Saccharomyces cerevisiae for production of ginsenosides.Metab Eng,2013,20:146-156.
����[88] Zhang GL,Cao Q,Liu JZ,et al.Refactoringβ-amyrin synthesis in Saccharomyces cerevisiae.AICh E J,2015,61(10):3172-3179.
����[89] Park SH,Hahn JS.Development of an efficient cytosolic isobutanol production pathway in Saccharomyces cerevisiae by optimizing copy numbers and expression of the pathway genes based on the toxic effect ofα-acetolactate.Sci Rep,2019,9:3996.
����[90] Zeng WZ,Guo LK,Xu S,et al.High-throughput screening technology in industrial biotechnology.Trends in Biotechnol,2020,38(8):888-906.
����[91] Rugbjerg P,Sommer MOA.Overcoming genetic heterogeneity in industrial fermentations.Nat Biotechnol,2019,37(8):869-876.
����[92] Wehrs M,Tanjore D,Eng T,et al.Engineering robust production microbes for large-scale cultivation.Trends Microbiol,2019,27(6):524-537.
����[93] Si T,Chao R,Min YH,et al.Automated multiplex genome-scale engineering in yeast.Nat Commun,2017,8:15187.
����[94] Wang HH,Isaacs FJ,Carr PA,et al.Programming cells by multiplex genome engineering and accelerated evolution.Nature,2009,460(7257):894-898.
����[95] Di Carlo JE,Conley AJ,Penttil?M,et al.Yeast oligo-mediated genome engineering (YOGE).ACSSynth Biol,2013,2(12):741-749.
����[96] Barbieri EM,Muir P,Akhuetie-Oni BO,et al.Precise editing at DNA replication forks enables multiplex genome engineering in eukaryotes.Cell,2017,171(6):1453-1467.e1413.
����[97] Jako?i��nas T,Rajkumar AS,Zhang J,et al.Cas EMBLR:Cas9-facilitated multiloci genomic integration of in vivo assembled DNA parts in Saccharomyces cerevisiae.ACS Synth Biol,2015,4(11):1226-1234.
����[98] Liu RM,Liang LY,Choudhury A,et al.Multiplex navigation of global regulatory networks (MINR) in yeast for improved ethanol tolerance and production.Metab Eng,2018,51:50-58.
����[99] Guo XG,Chavez A,Tung A,et al.High-throughput creation and functional profiling of DNA sequence variant libraries using CRISPR-Cas9 in yeast.Nat Biotechnol,2018,36(6):540-546.
����[100] Annaluru N,Muller H,Mitchell LA,et al.Total synthesis of a functional designer eukaryotic chromosome.Science,2014,344(6179):55-58.
����[101] Pennisi E.Building the ultimate yeast genome.Science,2014,343(6178):1426-1429.
����[102] Dymond JS,Richardson SM,Coombes CE,et al.Synthetic chromosome arms function in yeast and generate phenotypic persity by design.Nature,2011,477(7365):471-476.
����[103] Shen Y,Wang Y,Chen T,et al.Deep functional analysis of syn��,a 770-kilobase synthetic yeast chromosome.Science,2017,355(6329):eaaf4791.
����[104] Xie ZX,Li BZ,Mitchell LA,et al.“Perfect”designer chromosome��and behavior of a ring derivative.Science,2017,355(6329):eaaf4704.
����[105] Mitchell LA,Wang A,Stracquadanio G,et al.Synthesis,debugging,and effects of synthetic chromosome consolidation:syn��and beyond.Science,2017,355(6329):eaaf4831.
����[106] Wu Y,Li BZ,Zhao M,et al.Bug mapping and fitness testing of chemically synthesized chromosome��.Science,2017,355(6329):eaaf4706.
����[107] Zhang WM,Zhao GH,Luo ZQ,et al.Engineering the ribosomal DNA in a megabase synthetic chromosome.Science,2017,355(6329):eaaf3981.
����[108] Shao YY,Lu N,Wu ZF,et al.Creating a functional single-chromosome yeast.Nature,2018,560(7718):331-335.

���\���Ǵ��ϸ�����ĩ��ʼ������ۺ�ѧ�ơ�����Ϊ��21���Ϳ�ѧ�����ĺ���֮һ�������²��ϡ�����Դ���������Ӽ����γ���Ӱ��δ�������������Ĵ��ѧ��ϵ����ҽҩ����,���\���õ��˳���ķ�չ�����������ϡ�Ԥ�������������ش��ȷ��淢���˼����...