生物技术论文


“微生物群落”是地球上生命基本元素(C、N和S等)进行生物地球化学循环的主要驱动力,与人类健康、环境保护以及工农业生产等密切相关。近十年来,随着高通量测序的广泛应用“,微生物组学”成为新兴概念和热点。微生物组与不同的生存环境结合,诞生人体微生物组,宿主相关微生物组,一般环境微生物组,建筑环境微生物组,地球微生物组,医院环境微生物组等大量新兴的研究方向。
长期以来,研究方法一直是微生物群落研究的瓶颈。如,群落结构的阐述,即准确描述一定空间范围内的物种数量,并定量各物种的丰度,这是所有生态学研究的基本内容。然而,对微生物研究者而言,实现这一基本要求却绝非易事。这种困难主要源于微生物群落的如下几点特征。
(1)“微小”:宏观生物可以肉眼或者镜下观察其形态学分类特征并计数;而微生物即便在显微镜下也难以区分,形态差异特征少,因而不能直接观察种属并计数。
(2)“复杂”:微生物很少以纯种存在;但微生物群落含有极高的多样性。1 g土壤中可能含有数千到数万个不同种属的微生物。
(3)“稠密”:1 g土壤,1滴流水,都可能含有数以十亿计的微生物,并且它们常常来自成千上万的种属。
(4)“不均”:不同种属微生物在群落中的丰度差异极大。这种不均匀的分布特征造成优势种、非优势种以及稀有种的计数难以同时进行。
面对如此庞大复杂的微生物生态系统,微生物组学要准确理解样品中的微生物种类,多度及其功能,并将其与时间、空间、理化因素,宿主疾病状态等进行关联,从而探求微生物与微生物之间,微生物与宿主之间,以及微生物与环境之间的相互关系。因此,需要恰当的技术,在广度和精度这两个略显矛盾的角度,同时获得理想的数据。
自2006年,随着新一代高通量测序技术的成熟,不仅在人类基因组学领域带来了翻天覆地的变化,对微生物组学的研究产生了革命性的影响。当前,以16SrRNA高通量测序为基本手段,宏基因组鸟枪法测序、宏转录组、宏蛋白组、宏代谢组等组学领域产生了大量的新技术,共同促进了微生物组学的快速进步。
1 微生物组大数据分析的方法和流程
16S的测序是近年来微生物生态领域最核心、最重大的突破。通过454、Illumina等第2代测序仪高通量测定16S可变区序列,第1次让人们在可行的成本下,获得全面、系统、结构化的群落结构信息[1-2].美国WoodsHole 海洋研究实验室的 Mitchell Sogin 课题组于 2006年首次报道了通过焦磷酸测序技术,测定海洋沉积物样品的16S rRNA基因V6可变区,人类第1次在基本足够的测序深度下,清晰地展示了环境样品中微生物的组成,发现了高度的多样性。
与所有传统的微生物组学研究方法相比,该方法具有显着的优越性。该方法通过测定16S短片段序列,经生物信息学分析可以获得系统分类信息,从而可以明确定性其分类单元,不同实验间数据完全是可比较、可积累的。该方法通量显着提高,1次测定40~100万条序列,通过条码技术可以对每个样品测定数千到数万条短序列,从而可以获得广泛的、系统的结构信息。由于测序深度大,在多个数量级范围内可以进行定量。该方法的诞生对微生物组学的研究产生了巨大的影响,尤其对人体共生微生物领域最为活跃。例如,肥胖与部分肠道菌群间的相关性研究[3];人体不同部位的菌群结构的首次阐明[4];抗生素对肠道微生物群落产生的显着影响[5]等。在环境中,该技术首次在海洋沉积物中发现存在极其丰富、多样化的微生物群落。该方法让人们得以比较大空间尺度下土壤微生物群落结构的差异及其主要的影响因素(如pH)[6-8].
基 于 16S 的 分 析 可 称 为 宏 分 类 组 技 术(metataxonome)。16S的数据分析,其一般流程包括:序列提取、质控、相似序列聚类成OTU,种属分类,alpha以及beta多样性分析,以及进一步的统计分析。其中每一步都有关键之处,并正处于方法学前沿领域。
OTU聚类是16S序列分析的关键问题之一。在经典的分层聚类算法中,其运算量和所需的内存容量,均随着序列数量的增加呈几合级数增加。因此,贪婪算法成为目前该领域的主流。同时,也有不少研究者开发不基于序列比对的聚类算法。但是,由于序列相似性算法的不同,聚类中距离的传递问题,以及参考序列数据库的不足,该领域仍然存在运算效率和准确性问题。目前,与参比库比对的 Open- reference 算法[9]以及UPARSE是运用较为广泛的技术。
在完成聚类后,种属的分类仍然存在许多问题。目前,该领域主要通过与16S数据库比对,选取相似性高的参比序列的分类结果。但是,参比数据库本身,目前存在不少问题,例如目前应用最为广泛的Greengenes数据库[10],其中不少序列存在重复或者错误的分类结果。
UniFrac距离的计算,是beta多样性分析的关键工具。UniFrac距离是美国科罗达罗大学Rob Knight课题组创建的一种基于序列之间相似度,计算样品之间总的菌群距离的算法,有加权和不加权两种,在分析微生物群落相似性中均具有重要作用[11].基于UniFrac的工作基础,Rob Knight课题组进一步开发了微生物群落以及微生物生态分析的主流工具体系 QuantitiativeInsight Into the Microbial Ecology(QIIME)。该平台是一个流程的整合,已经在全球分析微生物组学科中广泛应用[12].
与之对应,Patrick Schloss开发了Mothur[13],该平台基于最初的序列聚类工具DOTUR而来。该平台和QIIME竞争,在许多地方有相似之处。二者之间的区别是,QIIME更为开放,系统整合能力更强,尊重方法的原创者,应用者更多一些,而Mothur则全部经作者改写,相对封闭。核糖体数据库RDP database课题组,也同样开发了针对二代测序数据的群落分析工具[14].除此之外,MG-RAST是一个综合性的在线数据分析平台[15].
使用者只需要将自己的测序数据投递到该网站,即可点击不同的宏基因组分析命令,完成数据分析。欧洲MetaHIT以及其它小组也开发了一些微生物群落的分析工具,但应用面不及上述几个平台。需要指出的是,除了16S外,人们还开发了一些针对特定功能基因的靶向测序技术,从而检测其功能多样性。其分析流程大体与16S相似,但需要特定的数据库加以比对分析。
宏基因组技术(metagenome),又称为元基因组技术,是在16S分析的基础上,通过宏基因组的鸟枪法高通量测序,能够同时获得菌群的分类信息以及功能基因的数据。并且该技术未经PCR扩增,因此PCR导致的偏差较少(测序建库时还会有部分PCR的影响)。因为微生物群落中不同微生物的多度差异极大,欲获得足够的定量信息,需要测试大量的数据。根据不同的需求,单个样品宏基因组测序的数据量,在Giga以上1~2个数量级水平。如此巨大的数据量,无论是测试成本,还是分析所需要消耗的机时,都相当可观。因此,人们通常在16S测试的基础上,挑选少量目标样品,测试其全基因组。当前,宏基因组数据的分析,通常包括如下步骤:
序列质控;将获得的高质序列组装(或者不经组装,直接与参比数据库比对);将组装后的序列与现有的微生物基因数据比对,并将比对上的序列进行门、纲、目、科、属、种的分类和丰度统计;进行样品间物种多样性的比较,如PCA分析、聚类分析、筛选与样品分组显着相关因子;进行基因组份分析,如前噬菌体预测、可转坐原件、基因预测;通过与KEGG、CAZy、eggNOG数据库比对进行功能注释,分析其中的代谢通路,碳水化合物活性酶、同源性;抗生素耐药组的比对分析等。在宏基因组分析中,针对病毒单独纯化的序列测序,可以获得病毒组数据,对微生物生态的解析,提供了全新的视野。
宏基因组测序和16S测序尽管在菌群分布上基本是一致的[17],但分辨效率显着不同。例如,在群落层面,二型糖尿病患者肠道菌群和对照人群并无显着的不同,但是,在宏基因组揭示的功能基因上,两组却呈现显着的差异[18-19].尽管宏基因组技术非常强大,该技术仍然存在诸多技术瓶颈。其一,大量序列目前尚无法找到匹配的数据库序列,尤其是病毒,大约80%甚至更多的序列无法注释;其二,仅仅通过序列相似度,对功能的注释常常是不准确的,存在大量的误注释;最后,对于大量的微生物基因组,通过宏基因组难以将其进行组装拼接,尤其是对低丰度的菌株。其中前两点缺陷同样适用于宏转录组学。
2 微生物组大数据分析的发展趋势
2.1 数据通量的进一步提高,成本的进一步下降
随着测序、质谱等技术的不断进步,依赖于上述技术的微生物组分析技术将同样不断升级换代。伴随着上述发展,解释微生物组所需要的数据量将不再成为瓶颈。多组学联合应用,将日益成为微生物组大数据分析的常用工具。
2.2 大数据分析成为领域的竞争焦点
当前,数据产生的效率,已经远远高于分析效率的提升。微生物组各类大数据的综合分析,日益成为瓶颈。如何储存,如何积累,如何提取关键信息,如何可视化展示,如何保证数据本身以及分析的可重现性,这一切都成为数据分析的挑战。同时,基于宏基因组数据的网络化分析和展示,是数据分析的新兴方向。
2.3 如何促进数据的标准化和可积累性日益显现
该问题初看不是科学问题,但却是限制我国相关学科发展的重要之处。如此庞大的数据量,如同迷宫一般的分析流程,不断升级的分析工具,许多原有的分析在不断纠错,这些数据的获得都来之不易,如何标准化,如何让后来者可以使用数据,这些对于微生物组学科学问题的解答至关重要。例如,我们发现,经典聚类算法直接导致了OTU的不稳定性,从而导致随着测序深度的变化[1],OTU的组成成员不断发生改变,进而影响到多样性的评估和差异物种的寻找,而该错误在大量的经典文献中都有所体现[20].有研究者正在探讨,运用iPython notebook工具,将原始数据的分析流程加以保存,从而重现全部的分析过程。
2.4 模型和预测是未来发展的趋势
随着数据量的日益增加,建立微生物生态模型,并预测微生物群落的动态发展,预测相关的生物学效应,是微生物组学研究的重要方向和关键应用。
2.5 数据知识产权保护以及数据本身价值的开发与保护
伴随着微生物组学大数据的不断发展,一个现实的问题是,分析工具网站以及数据储存网站,越来越倾向于要求使用者将原始数据以及研究相关的详细meta-data上传。这一方面使得数据的积累更加可靠。但同时,这种要求使得拥有数据库、掌握数据分析方法的团队,能够比实验者本身更早掌握研究的全面信息。这种大数据挖掘能力,令实验团队面临一定的知识产权损失风险。
2.6 培养和基于培养的功能验证将成为新的瓶颈需求
随着非培养技术提供越来越多的数据,如何验证基于大数据分析提出的假设,如何应用非培养提取出的重要菌株,这一切都需要获得纯培养菌株,在原位和实验室条件下验证其功能。
3 人体微生物组学发展趋势
尽管微生物组学呈现爆炸式发展趋势,该学科尚处在早期阶段,存在巨大的发展空间。作为人体生物医学研究的最后一块处女地(我们不能排除未来在核酸研究等领域存在新发现的可能),整个领域正处于从现象描述,关联分析,到机制研究,模型干预,最终到疾病诊断、预测和治疗的井喷式发展中。
随着数据的积累,人们将日益理解在复杂的人体微生物生态背后,哪些微生物是疾病或健康的驱动者,哪些是沉默者。基于此,我们将能理解何为正常菌群,何为失调菌群。人体微生物组在后天是如何形成、发展、稳定,以及与人体相互作用。从生态学角度,人体微生物组在不同部位形成以及改变的驱动因子是什么?
在应用角度,目前有两个重要的发展方向。其一,运用人体微生物组的组成或者功能基因,预测特定疾病的发生、发展以及结局。过去十多年,人们在运用人体基因组表达谱以及差异基因预测疾病和治疗效果上取得了长足的进步;可以预期,在未来的十年内,人们可以在微生物组上做出相似的工作。例如,人们发现IBD患者与非患者之间的菌群存在显着差异,可以将其作为非介入式疾病诊断的方法之一[21].通过对人体微生物组的干预和调整,治疗和预防疾病[22-23].与人体本身的基因组不同,人体微生物组是可以改变的。人们可以通过窄普抗生素、益生元、药物、饮食调整等策略,改善人体微生物生态。在此类研究中,宿主的选择性、生命早期事件、地区差异,肠道病毒组、人体微生物组之间的传递,以及人体微生物资源开发利用等,都存在大量的微生物生态学问题有待研究。我国传统医学在人体微生物生态方向积累了大量的经验,诸如祛湿、补脾健胃等都可能与人体菌群有关。如何通过现代人体微生物生态学手段,将传统经验进一步提取,澄清其背后的机制,开发药品和保健食品,保护好传统的知识产权,是我国相关科研人员的机遇与责任。
微生物组学研究,目的为回答谁、做什么,和怎么做三大基本问题。如果说过去方法学是核心瓶颈,如今条件则已基本具备。回答谁,人们可以采用16S和宏基因组测序,高通量培养,基因芯片,荧光原位杂交等技术。
回答做什么,人们可以采用宏转录组,宏蛋白组,宏代谢组,基因芯片,同位素标记,单细胞测序等手段。回答怎么做,则可以通过上述多组学与环境因子的关联数据挖掘,通过移植实验(例如无菌鼠验证肠道菌群的功能)等手段来解决。随着上述工具的建立,微生物组这一黑盒子正在打开。尽管高通量方法的成本依然偏高,但随着新工具的不断开发,微生物组分析的效率和准确性都将不断提高。如何利用好这些工具,关注微生物生态问题,而不是局限于追求一个完美的技术,值得国内研究者的重视。
参考文献:
[1]Sogin ML, Morrison HG, Huber JA, et al. Microbial diversity in thedeep sea and the underexplored “rare biosphere[''J]. Proc Natl AcadSci USA, 2006, 103(32): 12115-20.
[2]Huber JA, Mark Welch DB, Morrison HG, et al. Microbialpopulation structures in the deep Marine biosphere[J]. Science,2007, 318(5847): 97-100.
[3] Zhang H, Dibaise JK, Zuccolo A, et al. Human gut microbiota inobesity and after gastric bypass[J]. Proc Natl Acad Sci USA, 2009,106(7): 2365-70.
[4] Costello EK, Lauber CL, Hamady M, et al. Bacterial communityvariation in human body habitats across space and time[J]. Science,2009, 326(5960): 1694-7.
[5] Dethlefsen L, Huse S, Sogin ML, et al. The pervasive effects of anantibiotic on the human gut microbiota, as revealed by deep 16SrRNA sequencing[J]. PLoS Biol, 2008: e280.

水体富营养化主要是由于人为原因导致水体中氮、磷等营养元素远远超过水体的自净能力,从而在水体中富集,水体中的藻类因为有了丰富的营养物质得以急剧增长,细菌也大量繁殖。...

为了能够检测出食品中的微生物,相关工作人员将越来越多的新技术引入其中,以便能够做好食品检测工作,保障食品的安全性。...

随着污水处理厂的大规模增加,污水得到妥善的处理,而集中了大部分污染物的脱水后污泥的处置成了一个巫待解决的难题。传统的处理方式一般是填埋或焚烧,填埋势必造成长期的二次污染,使得污水处理的努力化为乌有,而焚烧会带来一些始料未及的环境污染。经过研究人...
近年来,组学技术(基因组学、转录组学、蛋白质组学和代谢组学)和基于组学的数学模型在生命科学领域变得越来越流行,而基于组学分析的系统发酵优化和遗传改造策略在产油微生物中也得到广泛应用。...
微生物能够产生多种具有生物活性的次级代谢产物,其中大部分活性次级代谢产物来源于放线菌[1],这些物质具有抗菌、抗肿瘤、杀虫、免疫抑制和免疫激活等功效,被广泛地应用于医药业、农业、兽业、食品工业等领域。近年来,随着抗生素耐药菌的不断出现,深入研...
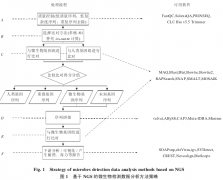
除了计算技术方面的优化,生物技术的创新也能给微生物检测方法带来革新。未来生物技术的发展使测序技术向着高通量、低成本、长读取长度的方向发展,目前已接近实用的第三代测序技术具有超长读长的特点,一旦应用将会极大地改善微生物检测方法的流程。对微生物...
淀粉是由脱水葡萄糖单位构成的生物高聚物,大部分是以淀粉颗粒的形态存在,不论来自何种植物或组织,都包含2种主要的多糖,即直链淀粉和支链淀粉。直链淀粉通常被定义为线性长链分子,是由脱水葡萄糖单位经过-1,4糖苷键连接而成的,而支链淀粉是由葡萄糖经...