医学遗传学论文

摘 要: 肌张力障碍是一种以持续性或间歇性肌肉收缩引起的异常运动和/或姿势为基本特征的运动障碍,具有重复性、模式化的特点,可被随意动作诱发或加重。遗传性因素所致的肌张力障碍即原发性肌张力障碍,目前共有28种已知表型。遗传学技术的发展极大地推进了遗传学机制的发现。尽管如此,仍有许多患者具有不同于这些表型的遗传学特点和临床特征。原发性肌张力障碍的诊断极具挑战性,需结合临床表现、影像学检查、肌电图、基因检测和其他临床检查综合判断。该文对原发性肌张力障碍的遗传学进展及诊断策略作一综述,旨在为临床实践及科学研究提供帮助。
关键词: 原发性肌张力障碍; 遗传学; 诊断;
Abstract: Dystonia is one kind of dyskinesia characterized by abnormal movement and/or posture caused by persistent or intermittent muscle contraction. It also has distinguished features of repeatability and modeling, and can be induced or aggravated by random movements. Dystonia caused by hereditary factors is named as primary dystonia. Currently, 28 phenotypes have already been found in primary dystonia. Development of genetic technology has largely promoted the discovery of genetic mechanisms. Even so, many patients still have different genetic and clinical features from these phenotypes. Diagnosis of primary dystonia is quite challenging. Clinical manifestations, imaging examinations, electromyography, gene testing and other examinations should be taken into account for systematic diagnosis. This article reviews the genetic progress and diagnostic strategies of primary dystonia, aimed at providing help for further clinical practice and scientific research.
Keyword: primary dystonia; genetics; diagnosis;、
肌张力障碍(dystonia,DYT)是一种以持续性或间歇性肌肉收缩引起的异常运动和/或姿势为基本特征的运动障碍,具有重复性、模式化的特点,可被随意动作诱发或加重[1]。根据有无遗传性病因,肌张力障碍可分为原发性和获得性。原发性肌张力障碍表型众多,目前主要可分为28种表型(DYT1~DYT21、DYT23~DYT29);其发病机制仍在探索中,而遗传学技术的快速发展不断揭示出各种肌张力障碍潜在的基因学异常。该类疾病的诊断尚无统一标准,不仅需考虑临床表现、影像学检查、肌电图和其他临床检查,遗传学特征也是重要的诊断及分型依据。本文详述原发性肌张力障碍的遗传学进展及诊断策略,以期为临床实践及科学研究提供新思路。
1 、遗传学进展
目前已有21种表型的相关遗传学位点被相继报道,但仍有7种表型尚未发现相关致病基因。此外,部分患者的表型及遗传学特点无法归类于上述28种表型之中。下文将对各型遗传学机制及其他相关基因进行总结。

1.1、 单纯性肌张力障碍
1.1.1、 DYT1
致病基因TOR1A(torsion-1A)编码的蛋白参与细胞脂质代谢、小脑突触连接的成熟及内质网应激。该蛋白C末端302/303位氨基酸位点缺失突变,导致ATP酶活性下调,从而影响上述生理过程[2,3]。
1.1.2、 DYT2
DYT2主要致病基因HPCA(hippocalcin)编码的海马钙结合蛋白属于神经元特异性钙结合蛋白家族成员,在脑部及视网膜具有钙离子感受器的功能。该基因突变致突触后抑制电位无法正常形成而致病[4]。
此外,Tuschl等[5]报道了SLC39A14(solute carrier family39,member 14)基因突变病例,表现为DYT2伴高锰血症(hypermanganesemia with dystonia-2,HMNDYT2)。该基因编码二价金属离子转运蛋白,参与锰的摄取与排泄。
1.1.3 、DYT4
致病基因TUBB4A(tubulinβ-4A)编码的蛋白属于脑特异性β-微管家族,主要表达于小脑、壳核及大脑白质。该基因突变可能导致微管网络紊乱、神经元及少突胶质细胞发育停滞,进而引起发病[6]。
1.1.4 、DYT6
致病基因THAP1(THAP domain-containing protein 1)编码的转录因子能调控自身表达,以及抑制TOR1A的表达,突变可干扰该抑制作用而致病。THAP1也参与其他神经元基因的转录调控过程[3,7]。
1.1.5 、DYT23
DYT23主要致病基因CACNA1B(voltagedependent N-type calcium channel subunitα-1B)编码N型电压依赖钙离子通道α-1B亚基,调控流入神经细胞内的钙电流以影响细胞膜兴奋性和抑制性突触递质的释放[8]。此外,CIZ1(CIP1-interacting zinc finger protein)基因突变也可致DYT23表型。CIZ1编码锌指蛋白1,与细胞周期蛋白依赖性激酶抑制剂p21存在相互作用,通过调控p21细胞内定位来促进DNA复制[9]。
1.1.6 、DYT24
致病基因ANO3(anoctamin-3)编码的蛋白属于钙激活氯离子通道蛋白家族,在壳核中高度表达,突变可致内质网钙信号降低[10,11]。
1.1.7、 DYT25
致病基因GNAL(guanine nucleotide-binding protein,α-activating activity polypeptide,olfactory type)编码Golf蛋白α亚基。Golf蛋白富集于纹状体,与DRD1(dopamine receptor 1)结合后,α亚基与β、γ亚基解离,后两者与GRK(G protein-coupled receptor kinase)结合引起下游效应器分子活动。GNAL基因突变可致β亚基与γ亚基结合力下降,Golf蛋白不稳定性增加,同时与多巴胺结合的DRD1对Golf蛋白的激动效应相应减弱[12]。
1.1.8、 DYT27
致病基因COL6A3(collagen typeⅥα-3chain)编码Ⅵ型胶原α3链,对维持细胞外基质的结构功能有重要作用。该基因突变可致异常的小脑-丘脑-皮质神经环路,或与轴突发育缺陷相关[13,14]。
1.1.9 、DYT28
致病基因KMT2B(lysine-specific methyltransferase2B)编码酪氨酸特异性甲基转移酶2B,对基因激活相关的表观遗传修饰十分关键。该基因突变影响组蛋白修饰和染色质状态,进而影响肌张力障碍相关基因的表达[15,16]。
1.1.10、其他相关基因
ATM(ataxia telangiectasia mutated gene)基因编码的蛋白属于磷脂酰肌醇3-激酶,参与DNA修复及细胞周期的调控。Necpal等[17]报道了1例携带ATM基因突变的不伴共济失调的节段性肌张力障碍患者,该患者12岁起病,以颅颈部及喉部受累为主;提示ATM与单纯性肌张力障碍存在一定相关性。
Mahajan等[18]报道了1例TPK1(thiamin kinase)基因突变的儿童期起病的单纯性全身性肌张力障碍患者。TPK1编码硫铵素焦磷酸激酶,能催化硫胺素转化为硫胺素焦磷酸酯,后者是参与多种糖代谢及能量代谢的活性因子。
1.2 、联合性肌张力障碍
1.2.1 、肌张力障碍叠加帕金森症状
(1)DYT3 DYT3的致病基因TAF1(TATA box-binding protein-associated factor 1)编码的TFⅡD(transcription factorⅡD)与TATA盒结合,构成DNA结合蛋白复合体,同RNA酶-Ⅱ9参与转录起始过程。神经元特异性TAF1缺失或为其致病机制[19]。
(2)DYT5、DYT14、SPR基因突变DYT5和DYT14肌张力障碍,以及SPR(sepiapterin reductase)基因突变引起的肌张力障碍均因体内多巴胺合成受影响而致病[20]。DYT5致病基因GCH1(GTP cyclohydrolase 1)编码GTP环水化酶1,是四氢生物蝶呤合成途径中合成多巴胺的限速酶。DYT14的致病基因TH(tyrosine hydroxylase)编码酪氨酸羟化酶,是酪氨酸转变为左旋多巴的关键酶。SPR基因编码蛋白则参与四氢生物蝶呤的合成过程。
(3)DYT12致病基因ATP1A3(sodium/potassiumtransporting ATPase subunitα-3)编码Na+-K+-ATP酶α3亚基,在大脑不同部位神经元特异性表达;突变通过影响蛋白活性或稳定性而致病[21]。正常情况下,该酶可通过消耗ATP以维持细胞内外钠钾离子浓度,维持静息电位。钠钙交换体则将钠离子转运至细胞内,将钙离子运至细胞外,维持细胞内钙浓度。当该酶功能受损,细胞内钠离子堆积,钠钙交换体反向转运,细胞内钙离子超载而致病[22]。
(4)DYT16 DYT16患者多因存在PRKRA(interferon-inducible double-stranded RNA-dependent protein kinase activator A)纯合突变c.665C>T/p.Pro222Leu而发病[23]。PRKRA编码蛋白激活因子PACT(PKR-activating protein)。在内质网应激或氧化应激时,PACT可致PKR(double-stranded RNA-activated protein kinase)磷酸化,激活eIF2a(eukaryotic translation initiation factor 2A),后者对下调蛋白质合成有重要作用。PRKRA基因突变可影响内质网应激和氧化应激,还可导致多巴胺信号转导通路、转录调控的异常[3]。
(5)其他相关基因Bris等[24]报道了1例NDUFS4(NADH-ubiquinone oxidoreductase Fe-S protein 4)基因突变的进展型多灶性肌张力障碍伴帕金森病的患者:12岁起病,初发为头部震颤及上肢肌挛缩,逐渐累及肩部、面部、喉部、呼吸肌等,伴显着的帕金森样症状。NDUFS4编码NADH氧化还原酶Fe-S蛋白4。该酶参与催化线粒体呼吸链,将电子从NADH传递至电子受体,对ATP的生成至关重要。
Rauschendorf等[25]曾报道2例SYNJ1(synaptojanin 1)基因突变的患者:初发为难治性癫痫,随后出现进展性全身性肌张力障碍伴舌部、头部和四肢的严重动作性震颤,多巴治疗有效;单光子发射计算机断层成像术(single photon emission computed tomography,SPECT)提示突触前多巴胺转运蛋白表达量下降,脑脊液检查则提示多巴胺合成缺陷。SYNJ1编码的突触小泡磷酸酶-1是一种聚磷酸肌醇磷酸酶,在被膜小窝和突触囊泡动力学中发挥作用,提示突触前囊泡功能缺陷所致的多巴胺合成障碍或为致病机制。
PSEN1(presenilin 1)基因突变可致常染色体显性遗传的阿尔茨海默病,但该基因同样与早发型肌张力障碍叠加帕金森病伴认知障碍相关。Carecchio等[26]报道了一个相关病例,并在神经病理上证实了黑质参与了发病。PSEN1编码早老素-1,是γ-分泌酶的催化部分,后者主导淀粉样蛋白前体和NOTCH受体蛋白的水解。
1.2.2、 肌张力障碍叠加肌阵挛
(1)DYT11 SGCE(epsilon-sarcoglycan)基因是DYT11主要的致病基因。携带来自父系的致病突变者发病,携带母系遗传的相同突变者往往无症状,或与SGCE基因母系印记有关。SGCE在不同部位表达不同剪切本,编码蛋白参与构成肌营养不良蛋白复合体。小脑特异性表达剪切本功能异常与该型有关[27]。
(2)DYT26致病基因KCTD17(potassium channel tetramerization domain-containing protein 17)编码钾离子通道四聚体结构域蛋白17。功能实验提示,在应激状态下患者的成纤维细胞质内钙信号转导受阻,内质网钙储存显着减少,或与疾病的发生相关[28]。
(3)其他相关基因Geiger等[29]报道了1例携带TUBB2B(tubulin beta-ⅡB)基因突变的肌阵挛-肌张力障碍患者:起病于16岁,磁共振成像(MRI)提示其脑部结构出现多重变异。TUBB2B编码的蛋白属于微管蛋白,突变或许通过影响细胞迁移而致病。
Balicza等[30]曾报道一家系,其中父亲和女儿均表现为伴运动发育迟缓、舞蹈病及垂体功能异常的肌阵挛-肌张力障碍,MRI提示空蝶鞍;NKX2-1(NK2 homeobox 1)为致病基因,其编码的转录因子对甲状腺、基底节、海马等前脑区域及肺的早期发育有重要的作用。
1.2.3、 发作性肌张力障碍叠加其他运动障碍
(1)DYT8致病基因MR1(myofibrillogenesis regulator 1)编码的蛋白参与神经突触调节,可按剪切位点分为3种亚型:MR-1L、MR-1M和MR-1S。MR-1L特异性表达于中枢神经系统,MR-1M和MR-1S则广泛表达于人体各组织。突变多位于MR-1L和MR-1S的N末端,因多巴胺能信号转导受干扰而致病[31]。
(2)DYT9、DYT18致病基因SLC2A1(solute carrier family 2,facilitated glucose transporter member 1)编码的蛋白为血管内皮细胞表达的葡萄糖转运蛋白,在中枢神经系统葡萄糖转运中发挥重要作用。突变可致血-脑屏障葡萄糖转运障碍,引起中枢神经系统能量代谢异常,引发癫痫性脑病[32]。
(3)DYT10 PRRT2(proline-rich transmembrane protein 2)是DYT10主要致病基因。PRRT2在中枢神经系统高表达,与SNAP25(synaptosomal-associated protein25)存在相互作用。SNAP25为可溶性SNARE(soluble NSF-attachment protein receptor)突触前蛋白,在基底节高表达,参与SNARE复合体的构成及神经突触囊泡的胞吐,在兴奋的神经元中负向调控电压门控性钙通道。该基因突变可能引发神经元兴奋性增高而致病[33]。
(4)其他相关基因ECHS1基因编码短链烯酰辅酶水合酶,催化线粒体脂肪酸β-氧化的第二步,同时也参与异亮氨酸及缬氨酸代谢过程,其突变可能通过影响线粒体代谢而致病。该型表现与发作性过度运动诱发性肌张力障碍类似,头颅MRI中苍白球高信号是其特征性表现[34,35]。
1.3 、复杂性肌张力障碍
1.3.1 、DYT29
DYT29呈常染色体隐性遗传,儿童期起病,主要表现为伴视神经萎缩和基底神经节异常的肌张力障碍。其疾病特征与线粒体遗传疾病相似,主要影响视神经、基底节等高能量需求和高氧化应激敏感度的部位,但患者认知功能相对保留,线粒体生物学标志物无明显异常。MECR(enoyl-[acyl-carrier-protein]reductase,mitochondrial)为其致病基因,编码线粒体反式-2-烯酰辅酶A还原酶,参与线粒体脂肪酸合成的最后一步,对线粒体呼吸链也有重要作用[36]。
1.3.2、 其他相关基因
Ortega-Suero等[37]曾报道1例OPA1(optic atropnhy 1 protein)基因突变的患者:40岁起病,初发为视神经萎缩相关表现,随后出现头颈部肌张力障碍。OPA1基因与线粒体的稳定性和能量代谢密切相关,突变则会影响眼、肌肉、神经等高能量需求的部位。
Rossi等[38]报道了1例POLG(polymerse DNA gamma)基因突变的患者:青少年起病,表现为合并全身性肌张力障碍的进行性眼外肌麻痹。POLG编码线粒体DNA多聚酶γ,突变的POLG表达的蛋白是该酶的竞争性抑制剂,可致线粒体DNA的点突变和/或缺失或致其拷贝数减少。
Radha Rama Devi等[39]报道了2例SERAC1(serine active site-containing protein 1)基因突变的患者:表现为进展型全身性肌张力障碍伴智力障碍、甲基丙二酸血症,MRI提示Leigh样病变。SERAC1编码蛋白主要参与磷脂酰甘油的重构,对线粒体功能和细胞内胆固醇运输至关重要。
1.4 、小结
原发性肌张力障碍具有极大的遗传异质性,目前已知的各表型遗传学及临床特征可见表1。此外,仍有部分表型的遗传学病因尚无定论,对这些空白领域的探索将有助于进一步了解该病的发病机制。
表1 原发性肌张力障碍的遗传学及临床特征
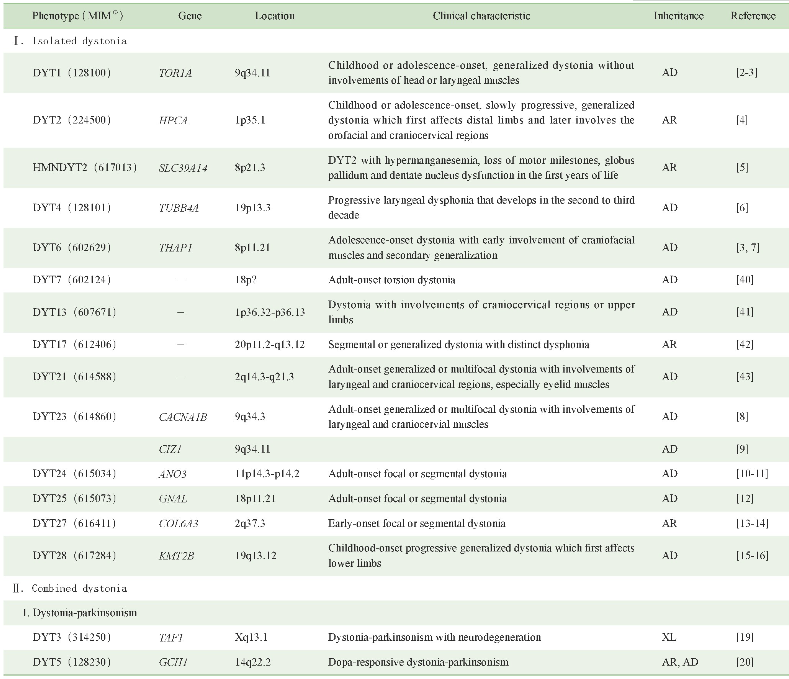
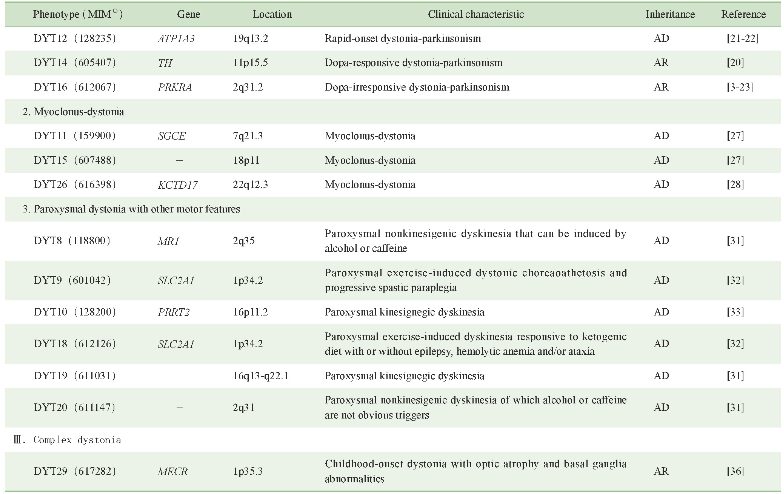
Note:(1)MIM number is a numerical assignment for inherited diseases,genes and functional segments of DNA,as listed in the comprehensive catalog Mendelian Inheritance in Man.“-”indicates that no corresponding pathogenic gene has been found.AD—autosomal dominant inheritance.AR—autosomal recessive inheritance.XL—X-linked inheritance.
2、 诊断策略
原发性肌张力障碍种类繁多,诊断极具挑战性,临床实践中可采用以下思路予以诊断[44]。
2.1、 明确是否为肌张力障碍
肌张力障碍的诊断以临床为主。其运动症状表现各异,但具有异常姿势、异常运动、感觉诡计、镜像现象及泛化现象等共同特征[45]。此外,肌电图可直观展示肌肉活动特点,影像学检查有助于排除继发性或症状性肌张力障碍,遗传学技术则可为诊断分型提供新依据。肌张力障碍常与其他运动功能亢进疾病相混淆;但其异常运动具有持续性、重复性及模式化,因而可与肌阵挛的单一电击样的抽动收缩,舞蹈症快速、非持续的不自主动作,震颤节律性、震荡性的不自主运动相鉴别。
2.2 、明确是否存在继发性因素
常见的继发性因素包括各类获得性脑损伤,以脑瘫最为常见。多巴胺阻滞剂可引发急性肌张力障碍,而迟发的药物性肌张力障碍则发生在服用各类神经松弛剂数天到数年后。此外,精神因素也是引发此病的继发性因素之一[46]。
2.3、 明确是否存在其他运动障碍表现
若无其他运动障碍表现,则支持单纯性肌张力障碍分类。若有其他运动障碍表现,则应:(1)确定主要运动障碍表现。(2)判断患者是否存在其他非运动症状。(3)判断患者是否存在其他神经系统异常及是否存在其他系统受累。(4)分析患者病情进展(发病年龄、起病速度和受累部位发展顺序)。(5)关注其他辅助检查。若患者的疾病特征与已知表型均不符,可考虑行全基因组测序,探究是否存在新基因突变。
2.4、 小结
目前,原发性肌张力障碍的诊断仍主要依赖于临床手段,分子遗传学技术则可在对患者临床分析的基础上,对疾病进行快速诊断分型。依据上述诊断流程(图1)进行全面的判断,将对治疗、预后、遗传咨询具有重要的参考价值。
图1 原发性肌张力障碍的诊断流程图
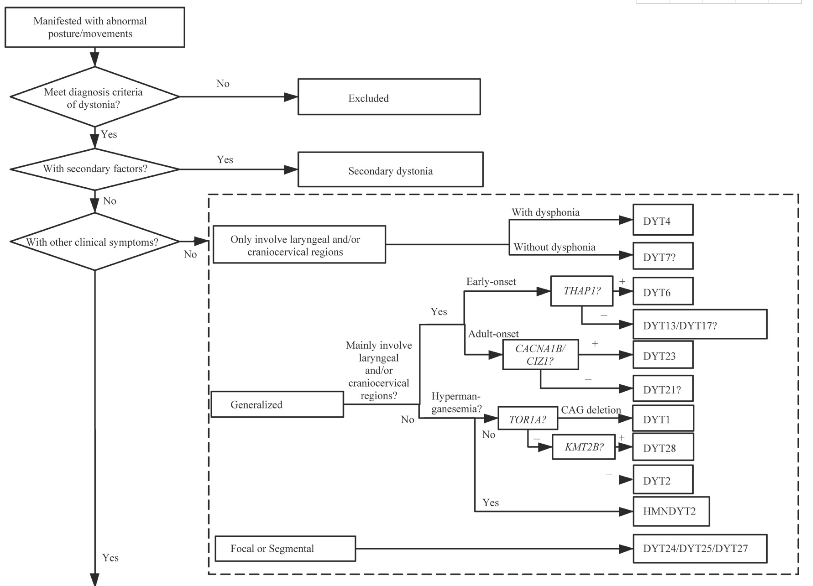
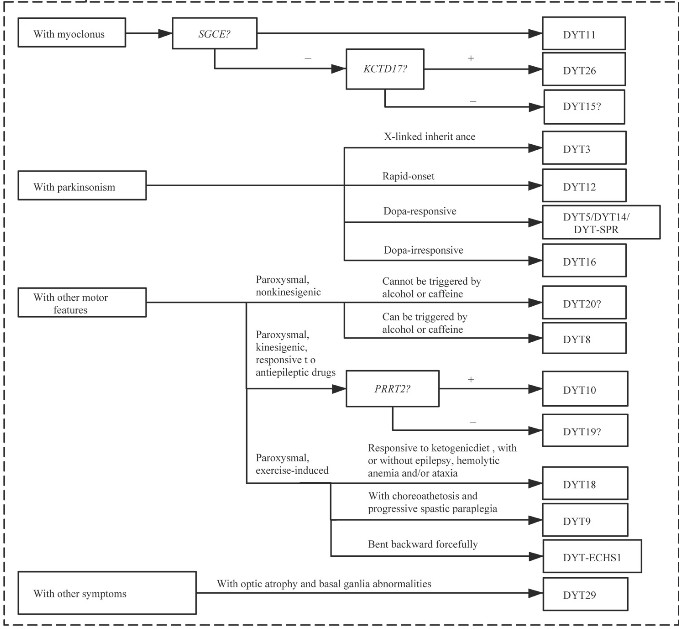
Fig 1 Diagnosis flow chart of primary dystonia
Note:As no corresponding pathogenic gene has been found for DYT7,DYT13,DYT15,DYT17,DYT19,DYT20,and DYT21,they cannot be diagnosed definitely by genetic test.DYT-SPR means dystonia with SPR gene mutation.DYT-ECHS1 means dystonia with ECHS1 gene mutation.
3 、结语
肌张力障碍是一类神经系统常见疾病;其中,遗传因素所致的肌张力障碍被称为原发性肌张力障碍,其诊断极具挑战性。随着分子遗传学技术的快速发展,我们对其遗传学机制的认识不断加深,极大地提高了对该类疾病的诊断、治疗水平;但其具体致病机制、诊治方法等仍存在许多问题,有待进一步探索和解决。
参考文献
[1]Albanese A,Bhatia K,Bressman SB,et al.Phenomenology and classification of dystonia:a consensus update[J].Mov Disord,2013,28(7):863-873.
[2]Goodchild RE,Buchwalter AL,Naismith TV,et al.Access of torsin A to the inner nuclear membrane is activity dependent and regulated in the endoplasmic reticulum[J].J Cell Sci,2015,128(15):2854-2865.
[3]Bragg DC,Armata IA,Nery FC,et al.Molecular pathways in dystonia[J].Neurobiol Dis,2011,42(2):136-147.
[4]Charlesworth G,Angelova PR,Bartolome-Robledo F,et al.Mutations in HPCAcause autosomal-recessive primary isolated dystonia[J].Am J Hum Genet,2015,96(4):657-665.
[5]Tuschl K,Meyer E,Valdivia LE,et al.Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia[J].Nat Commun,2016,7:11601.
[6]Watanabe N,Itakaoka M,Seki Y,et al.Dystonia-4(DYT4)-associated TUBB4Amutants exhibit disorganized microtubule networks and inhibit neuronal process growth[J].Biochem Biophys Res Commun,2018,495(1):346-352.
[7]Dauer W.Inherited isolated dystonia:clinical genetics and gene function[J].Neurotherapeutics,2014,11(4):807-816.
[8]Groen JL,Andrade A,Ritz K,et al.CACNA1B mutation is linked to unique myoclonus-dystonia syndrome[J].Hum Mol Genet,2015,24(4):987-993.
[9]Xiao J,Uitti RJ,Zhao Y,et al.Mutations in CIZ1 cause adult onset primary cervical dystonia[J].Ann Neurol,2012,71(4):458-469.
[10]Zech M,Boesch S,Jochim A,et al.Clinical exome sequencing in early-onset generalized dystonia and large-scale resequencing follow-up[J].Mov Disord,2017,32(4):549-559.
[11]Charlesworth G,Plagnol V,Holmstrom KM,et al.Mutations in ANO3 cause dominant craniocervical dystonia:ion channel implicated in pathogenesis[J].Am J Hum Genet,2012,91(6):1041-1050.
[12]Masuho I,Fang M,Geng C,et al.Homozygous GNAL mutation associated with familial childhood-onset generalized dystonia[J].Neurol Genet,2016,2(3):e78.
[13]Jochim A,Zech M,Gora-Stahlberg G,et al.The clinical phenotype of earlyonset isolated dystonia caused by recessive COL6A3 mutations(DYT27)[J].Mov Disord,2016,31(5):747-750.
[14]Jochim A,Li Y,Zech M,et al.Microstructural white matter abnormalities in patients with COL6A3 mutations(DYT27 dystonia)[J].Parkinsonism Relat Disord,2018,46:74-78.
[15]Zech M,Boesch S,Maier EM,et al.Haploinsufficiency of KMT2B,encoding the lysine-specific histone methyltransferase 2B,results in early-onset generalized dystonia[J].Am J Hum Genet,2016,99(6):1377-1387.
[16]Meyer E,Carss KJ,Rankin J,et al.Mutations in the histone methyltransferase gene KMT2B cause complex early-onset dystonia[J].Nat Genet,2017,49(2):223-237.
[17]Necpal J,Zech M,Skorvanek M,et al.Ataxia telangiectasia gene mutation in isolated segmental dystonia without ataxia and telangiectasia[J].Mov Disord Clin Pract,2018,5(1):89-91.
[18]Mahajan A,Sidiropoulos C.TPK1 mutation induced childhood onset idiopathic generalized dystonia:report of a rare mutation and effect of deep brain stimulation[J].J Neurol Sci,2017,376:42-43.
[19]Makino S,Kaji R,Ando S,et al.Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism[J].Am J Hum Genet,2007,80(3):393-406.
[20]Tadic V,Kasten M,Bruggemann N,et al.Dopa-responsive dystonia revisited:diagnostic delay,residual signs,and nonmotor signs[J].Arch Neurol,2012,69(12):1558-1562.
[21]Rosewich H,Thiele H,Ohlenbusch A,et al.Heterozygous de-novo mutations in ATP1A3 in patients with alternating hemiplegia of childhood:a whole-exome sequencing gene-identification study[J].Lancet Neurol,2012,11(9):764-773.
[22]Heinzen EL,Arzimanoglou A,Brashear A,et al.Distinct neurological disorders with ATP1A3 mutations[J].Lancet Neurol,2014,13(5):503-514.
[23]Quadri M,Olgiati S,Sensi M,et al.PRKRA mutation causing early-onset generalized dystonia-parkinsonism(DYT16)in an Italian family[J].Mov Disord,2016,31(5):765-767.
[24]Bris C,Rouaud T,Desquiret-Dumas V,et al.Novel NDUFS4 gene mutation in an atypical late-onset mitochondrial form of multifocal dystonia[J].Neurol Genet,2017,3(6):e205.
[25]Rauschendorf MA,Jost M,Stock F,et al.Novel compound heterozygous synaptojanin-1 mutation causes L-dopa-responsive dystonia-parkinsonism syndrome[J].Mov Disord,2017,32(3):478-480.
[26]Carecchio M,Picillo M,Valletta L,et al.Rare causes of early-onset dystoniaparkinsonism with cognitive impairment:a de novo PSEN-1 mutation[J].Neurogenetics,2017,18(3):175-178.
[27]Rachad L,El Kadmiri N,Slassi I,et al.Genetic aspects of myoclonus-dystonia syndrome(MDS)[J].Mol Neurobiol,2017,54(2):939-942.
[28]Mencacci NE,Rubio-Agusti I,Zdebik A,et al.A missense mutation in KCTD17 causes autosomal dominant myoclonus-dystonia[J].Am J Hum Genet,2015,96(6):938-947.
[29]Geiger JT,Schindler AB,Blauwendraat C,et al.TUBB2B mutation in an adult patient with myoclonus-dystonia[J].Case Rep Neurol,2017,9(2):216-221.
[30]Balicza P,Grosz Z,Molnar V,et al.NKX2-1 new mutation associated with myoclonus,dystonia,and pituitary involvement[J].Front Genet,2018,9:335.
[31]Gardiner AR,Jaffer F,Dale RC,et al.The clinical and genetic heterogeneity of paroxysmal dyskinesias[J].Brain,2015,138(Pt 12):3567-3580.
[32]Meneret A,Roze E.Paroxysmal movement disorders:an update[J].Rev Neurol(Paris),2016,172(8-9):433-445.
[33]Corradini I,Donzelli A,Antonucci F,et al.Epileptiform activity and cognitive deficits in SNAP-25+/-mice are normalized by antiepileptic drugs[J].Cereb Cortex,2014,24(2):364-376.
[34]Mahajan A,Constantinou J,Sidiropoulos C.ECHS1 deficiency-associated paroxysmal exercise-induced dyskinesias:case presentation and initial benefit of intervention[J].J Neurol,2017,264(1):185-187.
[35]Olgiati S,Skorvanek M,Quadri M,et al.Paroxysmal exercise-induced dystonia within the phenotypic spectrum of ECHS1 deficiency[J].Mov Disord,2016,31(7):1041-1048.
[36]Heimer G,Keratar JM,Riley LG,et al.MECR mutations cause childhood-onset dystonia and optic atrophy,a mitochondrial fatty acid synthesis disorder[J].Am J Hum Genet,2016,99(6):1229-1244.
[37]Ortega-Suero G,Fernandez-Matarrubia M,Lopez-Valdes E,et al.A novel missense OPA1 mutation in a patient with dominant optic atrophy and cervical dystonia[J].Mov Disord Clin Pract,2019,6(2):171-173.
[38]Rossi M,Medina Escobar A,Radrizzani M,et al.Dystonia in a patient with autosomal-dominant progressive external ophthalmoplegia type 1 caused by mutation in the POLG gene[J].Mov Disord Clin Pract,2017,4(2):266-269.
[39]Radha Rama Devi A,Lingappa L.Novel mutations in SERAC1 gene in two Indian patients presenting with dystonia and intellectual disability[J].Eur J Med Genet,2018,61(2):100-103.
[40]Klein C,Lohmann K,Marras C,et al.Hereditary dystonia overview[M/OL]//Adam MP,Ardinger HH,Pagon RA,et al.Gene Reviews.Seattle:University of Washington,2017.[2019-06-05].https://www.ncbi.nlm.nih.gov/books/NBK1155/.
[41]Leube B,Rudnicki D,Ratzlaff T,et al.Idiopathic torsion dystonia:assignment of a gene to chromosome 18p in a German family with adult onset,autosomal dominant inheritance and purely focal distribution[J].Hum Mol Genet,1996,5(10):1673-1677.
[42]Valente EM,Bentivoglio AR,Cassetta E,et al.DYT13,a novel primary torsion dystonia locus,maps to chromosome 1p36.13-36.32 in an Italian family with cranial-cervical or upper limb onset[J].Ann Neurol,2001,49(3):362-366.
[43]Chouery E,Kfoury J,Delague V,et al.A novel locus for autosomal recessive primary torsion dystonia(DYT17)maps to 20p11.22-q13.12[J].Neurogenetics,2008,9(4):287-293.
[44]Norgren N,Mattson E,Forsgren L,et al.A high-penetrance form of lateonset torsion dystonia maps to a novel locus(DYT21)on chromosome 2q14.3-q21.3[J].Neurogenetics,2011,12(2):137-143.
[45]Fung VS,Jinnah HA,Bhatia K,et al.Assessment of patients with isolated or combined dystonia:an update on dystonia syndromes[J].Mov Disord,2013,28(7):889-898.
[46]Sanger TD,Chen D,Fehlings DL,et al.Definition and classification of hyperkinetic movements in childhood[J].Mov Disord,2010,25(11):1538-1549.

人类群体遗传学是研究人群遗传结构及其变化规律的科学。人类在长期进化过程中形成了诸多自身独特的生物学特征,如上眼睑皱褶、耳垂类型、鼻梁形状等。这些特征的形成既受遗传调控,也与环境密切相关。研究不同特征的出现率及基因频率,探讨不同民族或族群间...

为解决目前难题, 需降低测序费用, 拓展样本量, 提高全基因组测序技术, 补充新的分析方法 (如上位性分析法、混合线性模型法等) 或扩展至基因链以外 (如基因表达等) 的联合研究, 以推进身高分子遗传学领域研究的发展。...

随着新一代测序技术广泛应用, 胚胎诊断技术准确性及效率均有很大提高。本文通过文献复习, 综述PGD的新进展以及其在遗传性耳聋疾病的应用。...

现主要从重要和新发现的候选基因及其表达过程的调控步骤,如基因甲基化、非编码RNA对基因的调控环节等方面对非综合征性小耳畸形遗传学研究现状及进展作一综述。...

如何对典型的遗传伦理案例做出以恰如其分地解释,引导学生确立科学的医学伦理观,使其在未来的医务生涯中面对医学伦理困惑时能够做出正确选择,是当前医学遗传学课堂教学一项重要课题。...
临床遗传学是人类医学遗传学的一个重要组成部分,它起始于20世纪40年代,近几十年来取得了长足的进步。相较于医学遗传学对于遗传学基础知识和理论的侧重,临床遗传学则更注重于遗传病的临床应用实践,主要提供包括各种遗传病的诊断(染色体分析、生化遗...

人类的眼裂方向、上眼睑皱褶、耳垂型、额头发际是由单基因控制的遗传性状, 其中水平型眼裂方向对斜位、有上眼睑皱褶对无皱褶、有耳垂对无耳垂、尖型额头发际对平型是显性遗传性状。...
同性恋(homosexuality)是一种古已有之的社会学和生物学现象,指不管有无性关系,性吸引的对象都完全或主要为同性[1].同性恋不仅存在于人类中,从赫里福郡公牛到恒河猴,同性和双性性行为广泛存在[2-3].过去同性恋一直被认为是一种精神疾病,随着人们认知...
表观遗传学是研究除DNA序列变化外的其他机制引起的细胞表型和基因表达的可遗传的改变。表观遗传学现象涉及组蛋白修饰、DNA甲基化、RNAi、染色质重塑等。表观遗传机制主要在基因转录层面调控基因表达,最近新兴起的RNA表观遗传则以N6-甲基化腺嘌呤为标志,研...
河弧菌(Vibriofluvialis)是一种新发的食源性致病菌,1975年首次分离于巴林1名腹泻患者粪便中,也曾被称为F群弧菌(GroupFvibrios)和EF-6弧菌(EF-6vibrios)。河弧菌自然分布于温暖含盐的水体中,可通过污染海产品、淡水及海水水体得以传...