医学遗传学论文

摘 要: 遗传易感性是指基于个人遗传背景的多基因遗传病发病风险,即来源于父母一方或双方的特定遗传变异在某些情况下会诱发疾病。在特定疾病的发病机制中某些高外显率的遗传变异发挥重要作用,此类疾病通过患病家系分析即可定位疾病相关遗传变异;但另一些低外显率变异的作用则不明显,需要大规模患病人群分析来解析遗传机制。近年来,随着二代测序和多组学分析技术的发展和基因组数据的大量积累,癌症、代谢性疾病、心脑血管疾病和精神疾病等疾病遗传易感性研究中取得了显着进展,为疾病的早期筛查和诊断治疗提供了参考。
关键词: 遗传易感性; 分析方法; 基因型; 表型;
Abstract: Genetic susceptibility refers to the risk of developing polygenetic diseases based on personal genetic background, that is, specific genetic variation from one or both parents can induce disease in some cases. The genetic variations of high penetrance play important roles in the pathogenesis of specific diseases, in which associated genetic changes can be identified through pedigree analysis, while effects of these variations with low penetrance are hard to determine, requiring large-scale population analysis to investigate the responsible genetic mechanism. In recent years, with the development of second generation sequencing and multifactorial analysis techniques as well as the accumulation of genomic data, significant progress has been made in the study of genetic susceptibility to diseases such as cancer, metabolic diseases, cardiovascular and cerebrovascular diseases, and mental diseases,which provides a reference for early screening, diagnosis, and treatment of human diseases.
Keyword: Genetic susceptibility; Research method; Genotype; Phenotype;
随着人类基因组计划的完成和后基因组计划的开展,人们对于基因组变异和疾病的认识也越来越深入。人类基因组上的变异主要分为三种:单核苷酸变异(SNP)、小片段序列的插入或删除(Small Indel)和大片段的结构性变异(SV)。任何一种变异都可能会引起个体的表型或特征发生改变甚至引起疾病。
根据基因变异-疾病的关系可以将人类疾病分为三类。第一类是单基因遗传病(Monogenic disease),仅一对等位基因的改变就会引发疾病并可以遗传给后代[1],如血友病[2]和白化病[3]。虽然每种单基因遗传病的发病率很低,但由于种类众多(约5 000~8 000种),总发病率高达6%[1]。第二类是多基因疾病(Multigenic/polygenic disease),也称为复杂性疾病(Complex disease),是涉及两个或两个以上的基因且通常和多种环境因素共同作用导致的疾病[4],如肿瘤[5]和糖尿病[6]等。第三类为获得性基因病,主要是由病原微生物通过感染将其基因入侵到宿主,导致宿主基因组改变,如艾滋病、HPV感染及乙型肝炎等。
遗传易感性研究通常关注的是与人类疾病相关的基因组变异,主要研究方法有基于家系的连锁分析(Linkage analysis)、关联分析(包含候选基因关联分析(Candidate gene association study)、全基因组关联分析(Genome-wide association study)和基因集合关联分析(Gene set analysis))等。本文将介绍各种遗传易感性研究方法的历史、原理及进展。

1、 基于家系的连锁分析
连锁分析研究的理论基础是疾病家系中致病的基因或者染色体区域与疾病性状共分离(Co-segregation),因此连锁分析主要应用于单基因疾病研究。两个在基因组上位置相邻的基因座上的等位基因由于连锁而共同分离,因此更容易作为一个单位共同遗传给后代。因此,患病个体除携带致病性变异外,还会表现出携带与致病变异连锁的遗传标记位点(单核苷酸多态位点或微卫星序列)。通过分析患者中共享的遗传标记位点即可实现对于致病变异的定位[7]。由于来自一个家庭中父方或母方的共分离现象可能仅限于在此家庭内部,因此连锁分析必须有患病家庭的数据。基于家系的连锁分析有主要分为参数分析法、非参数分析法和核心家系法。
1.1、参数分析法
参数分析法(又称基于模型分析法)是家系研究的传统方法,主要通过最大似然法(Maximum-likelihood analysis)和LOD值[8]来检测待研究的家系的遗传模式是否符合一个已知影响表型的基因的遗传模式(Mode of inheritance,MOI)[8,9]。LOD值于1955年由Morton首次提出[10],计算的是在一个家系中,两个基因座按一定重组率(θ值)进行连锁遗传的可能性Lθ与不连锁遗传的可能性L0.5之比,体现为优势比的对数。公式如下:
Z(θ)=LOD=log10LθL0.5=log10(1?θ)NR×θR0.5(NR+R)
其中NR指不连锁的子代数量,R指连锁的子代数量。参数0.5指的是根据孟德尔的自由组合定律,在随机情况下任何两个完全不连锁的基因座都有50%的重组率。当LOD值≥3,即连锁的可能性与不连锁的可能性之比大于等于1 000∶1时,可以认为这两个基因座在该家系样本中是连锁的[8,11],对应的p值为小于等于0.05[12]。
1992年,Schellenberg等人应用该方法鉴定了位于14染色体的ApoE4与家族型阿尔兹海默病连锁[13]。随后多项研究表明,ApoE4会显着增加阿尔兹海默病的风险。携带一个ApoE4拷贝会增加2~3倍患病风险,而携带两个拷贝患病风险会增加12倍[14]。病理学研究结果表明,携带ApoE4人群的大脑中积聚着更多的β淀粉样蛋白斑块,会阻断神经元-神经元信号[15]。此外,应用参数方法鉴定的与疾病连锁的基因位点的例子还包括家族非髓性甲状腺癌[16]、家族型鼻咽癌[17]及冠心病[18]等。
1.2、 非参数分析法
基于模型的参数分析法需要预先获得所研究性状的遗传模式、涉及的等位基因数量及外显率,因此前期模型的错误预设对结果的影响非常大。为了解决参数分析法对预设参数极度敏感的难题,Weeks和Lange于1988年提出了连锁分析的另一种研究方法——非参数分析法(又称非基于模型分析法)[19],检测的是该家系的遗传模式是否背离了预期的自由组合模式[9],可以应用于无法预知疾病的遗传模式或无法获取足量的家庭成员数据的情况。非参数方法的原理是通过状态同源(IBS,identical by state)和血缘同源(IBD,identical by descent)来进行等位基因共占(Allele sharing methods)分析[20,21,22,23]。IBS指一段DNA片段的序列在两个或多个个体中完全一致;若该IBS片段是遗传自同一个共同祖先,则该片段是IBD。IBD的片段同时也是IBS的,但是IBS的片段不一定都是IBD,还可能是由于突变或重组导致序列完全一致。非参数方法通过患者同胞对(ASP,affected sib pairs),患者亲戚对(ARP,affected relative pairs)和家系患者(APM,affected-pedigree-member)[24,25]三种设计来检测患病家系中患病和未患病个体的基因位点相似的程度,并判断导致疾病的位点与测到的位点是否连锁。非参数方法的优点是研究对象相对容易收集,但检验效能相对较低且样本量要求较大。
一项研究在123对慢性淋巴细胞性甲状腺炎同胞对中,使用非参数ASP方法鉴定了基因组上与自身免疫疾病连锁的5q31-q33区域和与慢性淋巴细胞性甲状腺炎连锁的8q23-q24区域[19]。此外,应用ASP鉴定的疾病连锁位点有白血病[23]等;应用ARP鉴定的包括系统性红斑狼疮[26]和炎症性肠病[27]等疾病;应用APM的研究主要有阿尔茨海默病[28,29]等。
1.3、 基于核心家系的研究方法
复杂疾病遗传易感性的分析最常用的方法是在人群中随机选取的病例-对照法,但是由于样本人群可能受到年龄、性别、种族或地域等因素而造成人群分层[30],从而对研究结果造成影响。家庭成员之间的遗传背景相似,因此使用患者核心家系(Nuclear family)作为样本进行基于家系的病例-对照研究可以避免样本分层造成的影响[31,32]。核心家系(又称三体家系)由一对夫妻及其至少一名患病子女构成[33],常用的研究方法是家系传递连锁不平衡检验(Transmission disequilibrium test, TDT)[31]。TDT方法是在家系内进行相关分析,观察双亲(至少一个为杂合子)将与候选致病位点连锁的等位基因位点传递给患者子代的概率是否明显增高而呈现出连锁不平衡。与非参数分析方法相比,TDT方法可以研究位点与发病程度的关系及基因间的相互作用,比如应用TDT方法解决了胰岛素基因与IDDM是否相关的问题[31]。此外,由于核心家系只需包含一个患病子代,因此达到同样的检验效能所需的样本量比非参数分析法大大减少。但缺点是晚发型疾病的患者不易取得其双亲的数据,且部分携带疾病易感基因的个体因外显不全而易被错误定为不患病个体。因而许多研究者提出了改善TDT的方法,比如Curtis提出以不患病同胞作为对照[34]。
核心家系可以用来鉴定多种疾病相关的基因和位点。例如,一项欧洲系统性红斑狼疮家系研究选取了103例核心家系及C1q 基因的 5 个tag-SNP进行分析,发现C1qA和 C1qC 与系统性红斑狼疮显着相关[35]。此外,目前研究新生突变的主要方法是收集核心家系成员的全基因组测序数据,通过比较父母与子代基因组的差异来寻找新生突变[36]。这种研究策略要求测序覆盖度足够大(大于30X),来避免测序中的抽样误差[37]。2015年解密发育障碍研究计划(The Deciphering Developmental Disorders Study)收集了100 6个核心家系,鉴定了12个新生突变与个体生长发育疾病相关[38]。
基于家系的连锁分析方法需要大量、完整且准确的家系样本数据,在实际研究中具有较大的局限性,而且无法用于分析复杂疾病。随着二代测序技术的发展,研究者们开发了更有效的新方法来替代基于家系的连锁分析方法。
2 、关联分析研究方法
关联分析(Association study)是一种基于群体而非家系的研究方法,原理是通过分析在病例和对照中频率有显着差异的等位基因,来鉴定与疾病或性状相关的易感基因和位点。关联分析可以筛选与疾病或性状相关的位点,但两者的因果关系无法推测[39,40]。1996年Lander提出了常见变异导致常见疾病假说(Common Disease/Common Variant,CD/CV)[41]。该假说推测所有患有特定疾病或带有特定表型的个体的基因组均有来自于一个共同祖先的常见变异。这些变异对疾病和表型的作用表现为累加效应或倍乘效应。根据该假说,疾病易感性是来源于某些特定的常见变异位点,这些位点在患者人群中的频率显着高于未患病人群。该假说适用于2型糖尿病相关的PPARγ基因[42]和阿尔兹海默病相关的APOEε4基因[43]。目前常用的关联分析方法有候选基因关联分析和全基因组关联分析两种。
2.1、 候选基因关联分析
候选基因关联分析通过分析候选基因中的tag SNP或者候选SNP在病例样本和对照样本中等位基因的频率,筛选出在统计学意义上具有显着频率差异的位点作为与疾病相关的位点。候选基因关联分析最重要的是样本人群和候选基因的挑选。病例和对照人群应选择具有相似遗传背景的样本,这样频率差异显着的位点才确实与疾病相关。候选基因位点可以通过以下几个途径挑选:(1)首先根据被研究的疾病(性状)的发病机制,选择可能相关的基因;再挑选可能会调控该基因或者编码蛋白的SNP(通常为标记SNP,即tag SNP)[44,45];(2)通过全基因组关联分析筛选的与疾病相关的基因和位点,然后将这些候选基因位点在另外一个人群中进行验证[46];(3)将其他模式生物(如小鼠模型)的研究结果中与疾病相关的基因和位点作为候选基因,在人类疾病样本中研究其同源基因的关联性[47]。
最早使用候选基因关联分析法定位的疾病关联基因的例子是1983年基因APOC3(Apolipoprotein C3)上的位点被发现与高甘油三酯血症和动脉粥样硬化相关[48]。随后,Kim等人使用比较基因组学和染色体遗传方法发现了与人类和猪肥胖特性相关的基因[49]。通候选基因关联分析还在108例藏族人样本中发现了EDAR基因的位点与血氧饱和度及血小板数量相关,提示EDAR基因对藏族人的高海拔低氧环境适应有正向的贡献[50]。
2.2、 全基因组关联分析
全基因组关联分析(Genome-wide association study, GWAS)是在具有不同特性的人群中进行全基因组水平的关联分析的方法。与候选基因关联分析不同,全基因组关联分析针对的是所有的遗传位点。20世纪90年代末,全世界开始大规模构建生物样本数据库。世界各地的研究者都可以上传或获取数据库中的样本和基因组数据,使得样本收集的成本和难度大幅下降[51]。2003年完成的人类基因组单体型图(HapMap Project)计划[52]为全基因组关联分析研究使用的绝大多数SNP位点提供了单体型信息。基于以上因素及全基因组分型芯片技术的发展,全基因组关联分析成为了鉴定复杂疾病或性状研究中最常用的方法。
全基因组关联分析的经典步骤为:(1)发现与疾病或性状显着相关的位点;(2)在扩大样本中重复发现关联位点;(3)精细定位;(4)通过功能分析验证候选位点[53]。其中第一步发现候选位点为最关键的步骤。一个位点的致病风险通常用优势比(Odds Ratio)来代表,指的是携带该位点中一个等位基因的样本的患病概率和不携带该等位基因的患病概率的比值。当病例组样本中等位基因频率高于对照组样本时,优势比大于1,因此可以通过统计检验优势比是否显着大于1来筛选与疾病显着相关的位点及等位基因。当分析的SNP数量多达数十万个的情况下,一般选择5×10-8作为p值显着的阈值[54]。第二步扩大样本验证是为了去除第一步发现的假阳性位点,通常在更大规模的疾病-对照样本中将发现的相关位点进行候选基因关联分析[55]。此时由于只针少数的候选基因位点进行分析,所以P值阈值可以不受限制(通常为0.05或0.01),样本数也可以极大的增加。
全基因组关联分析目前已经得到了非常广泛的应用。根据NHGRI-EBI GWAS Catalog的统计,截止到2019年4月6日,已发表了3 923项全基因组关联分析研究并发现了134 705个关联性。
2.3 、基于关联分析的其他研究方法
关联分析通常是针对一组病例-对照样本进行逐个位点的卡方检验。为了发现更多的候选位点及微效基因位点,研究者基于单点单组样本关联分析又开发了多种研究方法,比如imputation、meta-analysis、上位效应研究(Epistasis)和基因集合关联分析(Gene set analysis,GSA)。
运用imputation,可以基于已知的基因型(如国际人类基因组单体型图计划[56]和千人基因组计划[57]来预测缺失位点的基因型,使得用于分析的位点数大大增加,从而实现对于易感性位点的精细定位。而meta-analysis则可以将多个关联分析的结果进行整合分析,保证微效位点的有效检出。
人们发现大多数数量性状受多个位点共同作用的复杂性状[58],因此发现更多的微效位点及位点之间的相互作用非常重要。上位效应指的是一个位点的作用取决于其他一个或多个位点的现象[59]。这种现象导致了这些位点对性状的贡献度背离了线性叠加模型[60]。通过该方法,可以检测出多个位点之间的相互作用及其对表型的影响。基因集合关联分析则通常选择与待研究疾病或性状相关的通路上的基因作为候选基因集合,关注的不是单个基因位点而是整个代谢通路或多个候选基因整体与疾病和性状的关联性[61]。这种方法可以发现疾病和性状遗传机制中的“多基因微效”效应,是目前鉴定复杂疾病的新方法之一。
2.4 、关联分析的应用
关联分析经常用来鉴定复杂疾病的易感基因。2005年一项发表在Science杂志的研究鉴定了一个在老年性黄斑变性的病例-对照中有显着的频率差异的CFH基因位点[62]。这是第一项应用全基因组关联分析进行疾病易感基因定位的研究。在此之后,全基因组关联分析就成为了鉴定疾病易感基因和复杂性状相关基因的主要方法。现在全基因组关联分析的策略是综合多个表型(表型组)数据,使用大样本人群队列进行分析,如使用了344 369例样本,发现了编码的15个常见多态和9个低频突变与人类身体脂肪分布特征显着相关[63]。
关联分析还可以用来分析候选基因位点与位点附近的基因的表达量的关联性,即表达数量性状基因座(Expression Quantitative Trait Loci,简称eQTL)。eQTL是把基因的表达量作为数量性状,研究候选位点与基因表达的关系。例如通过关联分析发现的一个eQTL基因座为SORT1[64]。随后通过功能实验发现SORT1与低密度脂蛋白的代谢相关,对心血管疾病治疗有重要意义[64,65,66]。
除了鉴定疾病易感基因,全基因组关联分析还可用于群体遗传学研究。2010年的一项研究将藏族人作为“病例”,与其有相似遗传背景的汉族人作为“对照”,通过全基因组关联分析发现了EPAS1基因上的位点的汉藏频率差异最显着,且这些位点在藏族人中与较低的血红蛋白浓度显着相关[67]。
3 、连锁分析和关联分析的比较
连锁分析和关联分析是两种有本质区别的研究方法,前者研究的是在一个家系中基因的遗传特性是否与疾病的传递相关,适用于家系中首位携带致病位点的个体与所收集到最年轻的个体相隔一代或几代的情况;而后者是基于样本群体的基因频率差异研究该基因是否与患病相关,可以用于分析大量无亲缘关系但拥有一个遥远的共同祖先的患病个体。基于家系的连锁分析只适合研究单基因遗传病[68],且鉴定的与疾病相关的区域较大,而关联分析在发现疾病的微效遗传机制时比连锁分析更为有效[69],通常定位的区域非常短,因此可以应用于复杂疾病的研究。
同时关联分析也具有许多局限性。首先由于关联分析的样本在一定程度上是随机选择的,因此对照样本与病例样本的遗传背景不一致的情况会造成位点等位基因频率差异与是否患病无关,而与样本人群遗传背景相关。其次,样本的性别、年龄、职业等因素也会对结果产生影响[30]。因此必须控制样本人群分层来排除假阳性。可采用如下方法:①尽量选择与患病人群相对同源的群体作为对照样本(如职业人群队列或核心家系未患病成员);②分析时将多个变量作为协变量来排除其对结果的影响;③对结果的p值进行校正(如Genomic control校正[70])(见图1)。
图1 关联分析和连锁分析原理及应用的比较
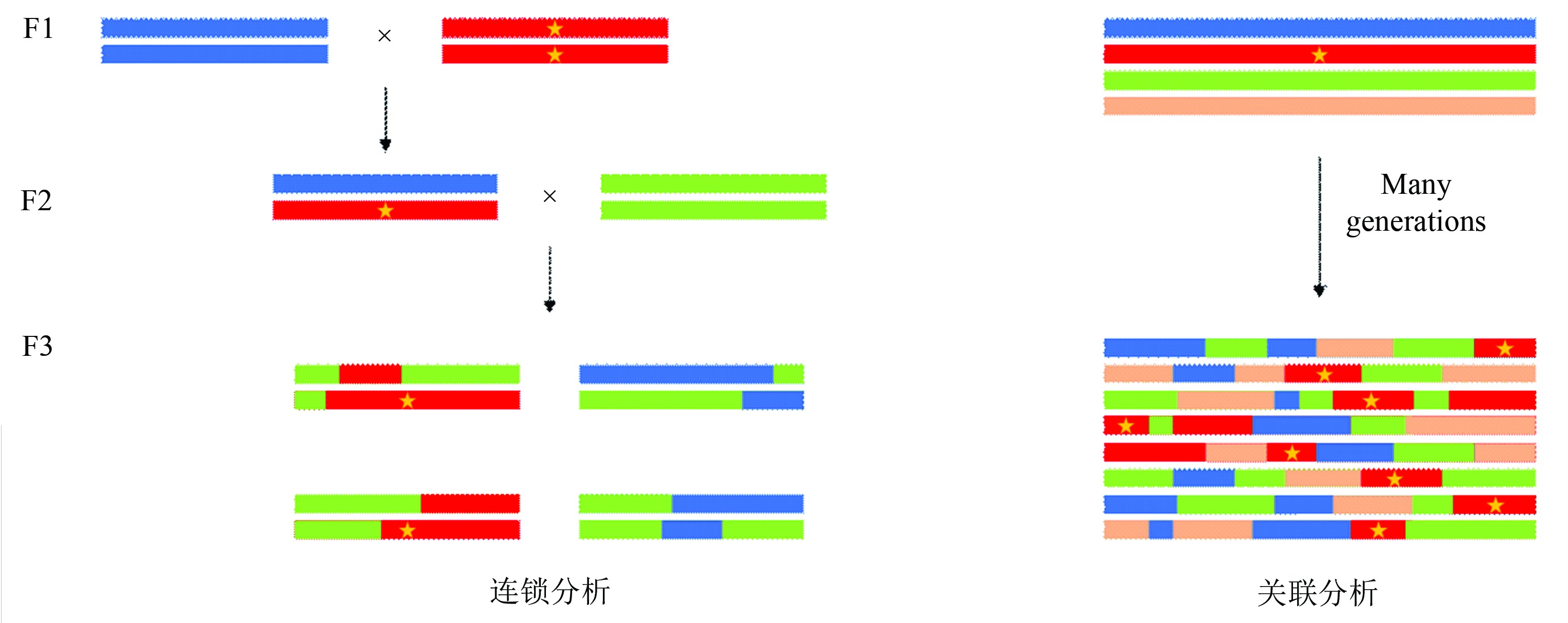
Fig.1 Linkage analysis and association mapping
4、 小 结
目前应用于疾病易感性研究的几种方法,这些方法均基于目前基因组分型和测序技术的快速发展及人类基因组数据的极大累积。每种方法均有各自的优势和局限性,因此研究者可以根据待研究的疾病或性状的特性及样本属性来选择合适的方法。由于疾病是多种环境和遗传因素共同作用的结果,发病机制非常复杂,因此未来仍需更多更有效的分析方法来研究基因组变异和疾病易感性的关系。
参考文献
[1]PRAKASH V,MOORE M,YANEZ-MUNOZ R J.Current progress in therapeutic gene editing for monogenic diseases[J].Molecular Therapy,2016,24(3):465-474.DOI:10.1038/mt.2016.5.
[2] ARONOVICH A,TCHORSH D,KATCHMAN H,et al.Correction of hemophilia as a proof of concept for treatment of monogenic diseases by fetal spleen transplantation[J].Proceedings of the National Academy of Sciences of the United States of America,2006,103(50):19075-19080.DOI:10.1073/pnas.0607012103.
[3] MEINDL A,HOSENFELD D,BR?CKL W,et al.Analysis of a terminal Xp22.3 deletion in a patient with six monogenic disorders:Implications for the mapping of X linked ocular albinism[J].Journal of Medical Genetics,1993,30(10):838-842.DOI:10.1136/jmg.30.10.838.
[4] GLAZIER A M,NADEAU J H,AITMAN T J.Finding genes that underlie complex traits[J].Science,2002,298(5602):2345-2349.DOI:10.1126/science.1076641.
[5]BALMAIN A.Cancer as a complex genetic trait:Tumor susceptibility in humans and mouse models[J].Cell,2002,108(2):145-152.DOI:10.1016/S0092-8674(02)00622-0.
[6]FIELD L L.Genetic linkage and association studies of Type I diabetes:Challenges and rewards[J].Diabetologia,2002,45(1):21-35.DOI:10.1007/s125-002-8241-7.
[7]BAILEY-WILSON J E,WILSON A F.Linkage analysis in the next-generation sequencing era[J].Human Heredity,2011,72:228-236.DOI:10.1159/000334381.
[8]GREENBERG D A,ABREU P,HODGE S E.The power to detect linkage in complex disease by means of simple LOD-score analyses[J].American Journal of Human Genetics,1998,63(3):870-879.DOI:10.1086/301997.
[9] KRUGLYAK L,DALY M J,REEVE-DALY M P,et al.Parametric and nonparametric linkage analysis:A unified multipoint approach[J].American Journal of Human Genetics,1996,58(6):1347-1363.
[10] MORTON N E.Sequential tests for the detection of linkage[J].American Journal of Human Genetics,1955,7(3):277-318.DOI:10.1016/S0065-2660(08)60097-8.
[11] RISCH N.Genetic-linkage:Interpreting lod scores[J].Science,1992,255(5046):803-804.DOI:10.1126/science.1536004.
[12] NYHOLT D R.All LODs are not created equal[J].American Journal of Human Genetics,2000,67(2):282-288.DOI:10.1086/303029.
[13] SCHELLENBERG G D,BIRD T D,WIJSMAN E M,et al.Genetic linkage evidence for a familial Alzheimer's disease locus on chromosome 14[J].Science,1992,258(5082):668-671.DOI:10.1126/science.1411576.
[14] CORDER E H,SAUNDERS A M,STRITTMATTER W J,et al.Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families[J].Science,1993,261(5123):921-923.DOI:10.1126/science.8346443.
[15] STRITTMATTER W J,SAUNDERS A M,SCHMECHEL D,et al.Apolipoprotein E:High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease[J].Proceedings of the National Academy of Sciences,1993,90(5):1977-1981.DOI:10.1073/pnas.90.5.1977.
[16] MCKAY J D,THOMPSON D,LESUEUR F,et al.Evidence for interaction between the TCO and NMTC1 loci in familial non-medullary thyroid cancer[J].Journal of Medical Genetics,2004,41(6):407-412.DOI:10.1136/jmg.2003.017350.
[17] XIONG W,ZENG Z Y,XIA J H,et al.A susceptibility locus at chromosome 3p21 linked to familial nasopharyngeal carcinoma[J].Cancer Research,2004,64(6):1972-1974.DOI:10.1158/0008-5472.Can-03-3253.
[18] LEPR?TRE F,BALGOBIN V,HEBE A,et al.A genome-wide scan for coronary heart disease suggests in Indo-Mauritians a susceptibility locus on chromosome 16p13 and replicates linkage with the metabolic syndrome on 3q27[J].Human Molecular Genetics,2001,10(24):2751-2765.DOI:10.1093/hmg/10.24.2751.
[19] TAMAI H,KUMA K,YAMAMOTO K,et al.Identification of susceptibility loci for autoimmune thyroid disease to 5q31-q33 and Hashimoto’s thyroiditis to 8q23-q24 by multipoint affected sib-pair linkage analysis in Japanese[J].Human Molecular Genetics,2001,10(13):1379-1386.DOI:10.1093/hmg/10.13.1379 .
[20] FISHMAN P M,SUAREZ B,HODGE S E,et al.A robust method for the detection of linkage in familial disease[J].American Journal of Human Genetics,1978,30(3):308-321.
[21] GREEN J R ,WOODROW J C.Sibling method for detecting HLA-linked genes in disease[J].Tissue Antigens,1977,9(1):31-35.DOI:10.1111/j.1399-0039.1977.tb01076.x.
[22] DAY N E,SIMONS M J.Disease susceptibility genes-their identification by multiple case family studies[J].Tissue Antigens,1976,8(2):109-119.DOI:10.1111/j.1399-0039.1976.tb00560.x.
[23] PENROSE L S.The detection of autosomal linkage in data which consist of pairs of brothers and sisters of unspecified parentage[J].Annals of Eugenics,1935,6(2):133-138.DOI:10.1111/j.1469-1809.1935.tb02224.x.
[24] AMOS C I,DAWSON D V ,ELSTON R C.The probabilistic determination of identity-by-descent sharing for pairs of relatives from pedigrees[J].American Journal of Human Genetics,1990,47(5):842-853.
[25] WEEKS D E,LANGE K.The affected-pedigree-member method of linkage analysis[J].American Journal of Human Genetics,1988,42(2):315-326.
[26]BAUTISTA J F,KELLY J A,HARLEY J B,et al.Addressing genetic heterogeneity in complex disease:Finding seizure genes in systemic lupus erythematosus[J].Epilepsia,2008,49(3):527-530.DOI:10.1111/j.1528-1167.2007.01453.x.
[27] DAVID A,VAN H,SHEILA A,et al.Inflammatory bowel disease susceptibility loci defined by genome scan meta-analysis of 1952 affected relative pairs[J].Human Molecular Genetics,2004,13(7):763-770.DOI:10.1093/hmg/ddh090.
[28]SAUNDERS A M,STRITTMATTER W J,SCHMECHEL D,et al.Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease[J].Neurology,1993,43(8):1467-1472.DOI:10.1212/WNL.43.8.1467.
[29] PERICAKVANCE M A,BEBOUT J L,GASKELL P C,et al.Linkage studies in familial alzheimer-disease-evidence for chromosome-19 linkage[J].American Journal of Human Genetics,1991,48(6):1034-1050.DOI:10.1016/0378-1119(91)90556-Q.
[30] NOVEMBRE J,JOHNSON T,BRYC K,et al.Genes mirror geography within Europe[J].Nature,2008,456(7218):98-101.DOI:10.1038/nature07331.
[31]BACANU S A,DEVLIN B ,ROEDER K.The power of genomic control[J].American Journal of Human Genetics,2000,66(6):1933-1944.DOI:10.1086/302929.
[32] SPIELMAN R S,MCGINNIS R E,EWENS W J.Transmission test for linkage disequilibrium- the insulin gene region and insulin-dependent diabetes-mellitus (Iddm)[J].American Journal of Human Genetics,1993,52:506-516.
[33]BENGTSON V L.Beyond the nuclear family:The increasing importance of multigenerational bonds.[J].Journal of Marriage and Family,2001,63(1):1-16.DOI:10.1111/j.1741-3737.2001.00001.x.
[34]CURTIS D.Use of siblings as controls in case-control association studies[J].Annals of Human Genetics,1997,61(pt4):319-333.DOI:10.1046/j.1469-1809.1998.6210089.x.
[35] MARTENS H A,ZUURMAN M W,DE LANGE A H M,et al.Analysis of C1q polymorphisms suggests association with systemic lupus erythematosus,serum C1q and CH50 levels and disease severity[J].Annals of the Rheumatic Diseases,2009,68(5):715-720.DOI:10.1136/ard.2007.085688.
[36] ACUNA-HIDALGO R,VELTMAN J A ,HOISCHEN A.New insights into the generation and role of de novo mutations in health and disease[J].Genome Biology,2016,17(1):241.DOI:10.1186/s13059-016-1110-1.
[37]ACUNA-HIDALGO R,BO T,MICHAEL,et al.Post-zygotic point mutations are an underrecognized source of de novo genomic variation[J].The American Journal of Human Genetics,2015,97(1):67-74.DOI:10.1016/j.ajhg.2015.05.008.
[38] STUDY T D D D,FITZGERALD T,GERETY S,et al.Large-scale discovery of novel genetic causes of developmental disorders[J].Nature,2015,519(7542):223.DOI:10.1038/nature14135.
[39] MANOLIO T A.Genomewide association studies and assessment of the risk of disease[J].New England Journal of Medicine,2010,363(2):166-176.DOI:10.1056/NEJMra0905980.
[40] PEARSON T A,MANOLIO T A.How to interpret a genome-wide association study[J].The Journal of the American Medical Association (JAMA),2008,299(11):1335-1344.DOI:10.1001/jama.299.11.1335.
[41] LANDER E S.The new genomics:Global views of biology[J].Science,1996,274(5287):536-539.DOI:10.1126/science.274.5287.536.
[42]REICH D E,LANDER E S.On the allelic spectrum of human disease[J].Trends in Genetics,2001,17(9):502-510.DOI:10.1016/S0168-9525(01)02410-6.
[43] SILL?N A,ANDRADE J,LILIUS L,et al.Expanded high-resolution genetic study of 109 Swedish families with Alzheimer’s disease[J].European Journal of Human Genetics,2007,16(2):202.DOI:10.1038/sj.ejhg.5201946.
[44] KWON J M,GOATE A M.The candidate gene approach[J].Alcohol Research Current Reviews,2000,24(3):164-168.DOI:10.1007/springerreference_34556.
[45] COLLINS F S,GUYER M S,CHAKRAVARTI A.Variations on a theme:Cataloging human DNA sequence variation[J].Science,1997,278(5343):1580-1581.DOI:10.1126/science.278.5343.1580.
[46] CHANG B L,CRAMER S D,WIKLUND F,et al.Fine mapping association study and functional analysis implicate a SNP in MSMB at 10q11 as a causal variant for prostate cancer risk[J].Human Molecular Genetics,2009,18(7):1368-1375.DOI:10.1093/hmg/ddp035.
[47]GRAHAM D S C,VYSE T J.The candidate gene approach:Have murine models informed the study of human SLE?[J].Clinical and Experimental Immunology,2004,137(1):1-7.DOI:10.1111/j.1365-2249.2004.02525.x.
[48] REES A,SHOULDERS C C,STOCKS J,et al.DNA polymorphism adjacent to human apoprotein A-1 gene:Relation to hypertriglyceridaemia[J].The Lancet,1983,321(8322):444-446.DOI:10.1016/S0140-6736(83)91440-X.
[49] KIM J,LEE T,KIM T-H,et al.An integrated approach of comparative genomics and heritability analysis of pig and human on obesity trait:Evidence for candidate genes on human chromosome 2[J].BMC Genomics,2012,13(1):711-711.DOI:10.1186/1471-2164-13-711.
[50]SHAO J,RAZA M S,ZHUOMA B,et al.Evolutionary significance of selected EDAR variants in Tibetan high-altitude adaptations[J].Science China Life Sciences,2018,61(1):68-78.DOI:10.1007/s11427-016-9045-7.
[51]GREELY H T.The uneasy ethical and legal underpinnings of large-scale genomic biobanks[J].Annual Review of Genomics and Human Genetics,2007,8(8):343-364.DOI:10.1146/annurev.genom.7.080505.115721.
[52] CONSORTIUM I H.The international HapMap project[J].Nature,2003,426(6968):789.DOI:10.1038/nature02168.
[53] IOANNIDIS J P A,THOMAS G,DALY M J.Validating,augmenting and refining genome-wide association signals[J].Nature Reviews Genetics,2009,10(5):318.DOI:10.1038/nrg2544.
[54] CLARKE G M,ANDERSON C A,PETTERSSON F H,et al.Basic statistical analysis in genetic case-control studies[J].Nature Protocols,2011,6(2):121-133.DOI:10.1038/nprot.2010.182.
[55]HOGGART C J,CLARK T G,DE IORIO M,et al.Genome-wide significance for dense SNP and resequencing data[J].Genetic Epidemiology:The Official Publication of the International Genetic Epidemiology Society,2008,32(2):179-185.DOI:10.1002/gepi.20292.
[56] GIBBS R A,BELMONT J W,HARDENBOL P,et al.The international HapMap project[J].Nature,2003,426(6968):789-796.DOI:10.1038/nature02168.
[57] CONSORTIUM G P.A map of human genome variation from population-scale sequencing[J].Nature,2010,467(7319):1061.
[58]VISSCHER P M,BROWN M A,MCCARTHY M I,et al.Five years of GWAS discovery[J].The American Journal of Human Genetics,2012,90(1):7-24.DOI:10.1016/j.ajhg.2011.11.029.
[59]GROS P-A,LE NAGARD H,TENAILLON O.The evolution of epistasis and its links with genetic robustness,complexity and drift in a phenotypic model of adaptation[J].Genetics,2009,182(1):277-293.DOI:10.1534/genetics.108.099127.
[60] WADE M J,GOODNIGHT C J.Cyto-nuclear epistasis:Two-locus random genetic drift in hermaphroditic and dioecious species[J].Evolution,2006,60(4):643-659.DOI:10.1554/05-019.1.
[61] DE LEEUW C A,NEALE B M,HESKES T,et al.The statistical properties of gene-set analysis[J].Nature Reviews Genetics,2016,17(6):353-364.DOI:10.1038/nrg.2016.29.
[62] KLEIN R J,ZEISS C,CHEW E Y,et al.Complement factor H polymorphism in age-related macular degeneration[J].Science,2005,308(5720):385-389.DOI:10.1016/s0084-392x(08)70394-0.
[63] JUSTICE A E,KARADERI T,HIGHLAND H M,et al.Protein-coding variants implicate novel genes related to lipid homeostasis contributing to body-fat distribution[J].Nature Genetics,2019,51:452-469.DOI:10.1038/s41588-018-0334-2.
[64] KATHIRESAN S,WILLER C J,PELOSO G M,et al.Common variants at 30 loci contribute to polygenic dyslipidemia[J].Nature Genetics,2009,41:56.DOI:10.1038/ng.291.
[65] BAUER R C,STYLIANOU I M,RADER D J.Functional validation of new pathways in lipoprotein metabolism identified by human genetics[J].Current Opinion in Lipidology,2011,22(2):123-128.DOI:10.1097/mol.0b013e32834469b3.
[66] DUB? J B,JOHANSEN C T,HEGELE R A.Sortilin:an unusual suspect in cholesterol metabolism:from GWAS identification to in vivo biochemical analyses,sortilin has been identified as a novel mediator of human lipoprotein metabolism[J].Bioessays,2011,33(6):430-437.DOI:10.1002/bies.201100003.
[67] BEALL C M,CAVALLERI G L,DENG L,et al.Natural selection on EPAS1 (HIF2α) associated with low hemoglobin concentration in Tibetan highlanders[J].Proceedings of the National Academy of Sciences of the United States of America,2010,107(2010):11459-11464.DOI:10.1073/pnas.1002443107.
[68]ALTM?LLER J,PALMER L J,FISCHER G,et al.Genomewide scans of complex human diseases:True linkage is hard to find[J].The American Journal of Human Genetics,2001,69(5):936-950.DOI:10.1086/324069.
[69] RISCH N,MERIKANGAS K.The future of genetic studies of complex human diseases[J].Science,1996,273(5281):1516-1517.DOI:10.1097/00001648-199805000-00023.
[70] DEVLIN B,ROEDER K.Genomic control for association studies[J].Biometric,1999,55(4):997-1004.DOI:10.1111/j.0006-341x.1999.00997.x.

p53基因第72密码子多态性与胃癌存在易感性。有报道认为,其易感性存在区域性[1-3]。青海地区地处青藏高原,平均海拔3500m,是胃癌高发区,胃癌检出率高达10.7%,高于其他地区[4]。本研究旨在通过对该地区胃癌基因型的分析,探讨p53基因Arg7...
现代许多临床研究已表明,人类血小板抗原(HPA)所导致的血小板同种免疫与新生儿同种免疫性血小板减少症(NATP)、血小板输注无效(PRT)、输血后紫癜(PTP)、移植相关血小板减少症、动脉血栓性疾病等相关[1,2].而不同人群中的HPA抗原的分布频率是与获得HP...