VIP 是 2016 年 Li[27]提出的微生物检测数据分析方法。VIP 其处理流程为:对于样本数据,首先进行去接头,低质量序列去除,低复杂度序列去除(环节A),得到 clean data.随后分为两种模式,牺牲精度换取速度的快速模式和注重精度的高敏感度模式。两种模式的区别体现在宿主基因组消减(环节C)环节。快速模式下,利用 Bowtie2 将 cleandata 与 MRG 进行比对,比对成功后直接输出结果。在高敏感度模式下,首先去除 clean data 中的细菌DNA 和全部 rRNA,剩余序列再利用 Bowtie2与MRG 进行比对 . 未比对成功的序列再利用RAPSearch 与 NCBI 的病毒蛋白质库进行比对。所有的比对成功的序列都进行de novo组装和系统发育分析(环节 F)。VIP 适用于高精度要求下的微生物检测问题。
3 “速度型”检测方法的安装配置及性能评估。
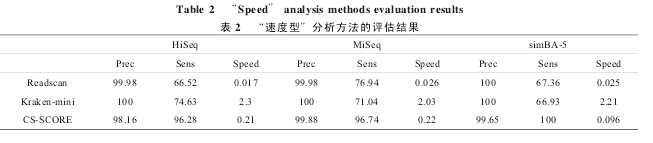
为了评估基于 NGS 的微生物检测数据分析方法,我们这里对“速度型”数据分析方法进行了安装配置与性能评估( 表 2),也就是: Readscan、Kraken 和 CS-SCORE,其中 Kraken 采用缩减数据库的Kraken-mini 版本。评估的内容包括:环境配置依赖、安装方法、计算速度、结果精度以及参考数据库比较等。测试环境为共享内存计算系统(Intel Xeon E7-8800 V3 CPU x 8、 1TB MEM、SSD、CentOS release 6.5 server)。
环境配置依赖和安装方法参见 github 项目
https://github.com/FreezeFish/MicrobeDetectionEvaluation.git. 项 目 给 出 了 Kraken-mini, CS-SCORE 和Readscan的数据下载,环境配置等自动化安装方法。
测试数据采用3个微生物宏基因组数据文件,分别称为“HiSeq”、“MiSeq”和“simBA-5”,来自于文献[21].根据文献[21],HiSeq和MiSeq序列数据来自两组不同的细菌宏基因组,均包含10个物种,通过鸟枪法测序得到。simBA-5宏基因数据由提高5倍突变率的细菌和古生物细菌序列混合构建。这 3 个序列文件都包含 10 000 000 个 reads,其中 HiSeq 平均读长为 92 bp,MiSeq 平均读长为156 bp,simBA-5 所有 reads 读长均为 100 bp.
计算速度采用总碱基量除以单次运行时间进行评估,单位采用Mbp/s,结果保留两位有效数字。
结果正确性采用精度(precision,prec)和敏感度(sensitivity,sens)两个指标进行评估。待测方法在运 行 完 成 后 均 会 给 每 条reads打 出 分 类 标 签(assignments),精度采用正确的标签占能够进行分类的标签的比例,敏感度指正确的标签占所有标签的比例。
从运行结果中可以看出,Kraken-mini 速度最快,CS-SCORE 其次,Readscan 运行速度较慢。并且随着reads 长度增加,Kraken-mini 的运行速度降低,但总体速度差别不大。Readscan 处理速度随着 reads 长度增加反而有所增加。CS-SCORE 运行速度和reads 长度没有确定关系。在结果正确性方面,Kraken-mini 对三个测试数据集都给出了100%的精度,这是因为 Kraken-mini 采用精确k-mer匹配,不考虑突变造成的影响。而 Readscan和CS-SCORE 则存在将微生物序列 reads 判别成人类序列 reads 的情况。Kraken-mini 的敏感度较差,是由于采用k-mer的比对方法,k-mer越小意味着特异性降低,和多条reference 比对成功的几率更高,而采用LDA 分类的方法只能向上一级进行分类,导致敏感度降低。
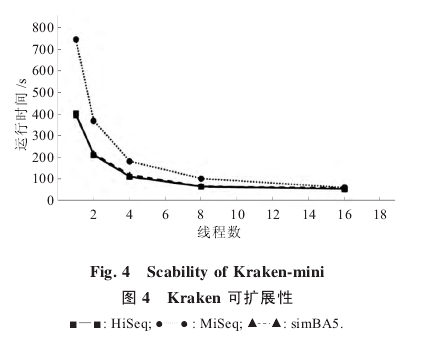
Kraken-mini 提供了多线程处理方法,处理各个样本数据结果如图4.
对于HiSeq 和 simBA5, Kraken 在 8 线程时能提供> 0.737 的加速比,但是到 16 线程加速比锐减到< 0.460.而 MiSeq 在 16 线程仍有 0.745 的加速比,由于MiSeq 数据量较大,结果合理。