司法鉴定论文


一、签字笔字迹形成时间检验研究综述
签字笔是目前被广泛使用的书写工具之一,它具有书写流利、 形成字迹清晰、 稳定等特点,人们经常使用其书写各类文件, 如信件、 合同、试卷、 收条欠条、 各类票据和表格等. 与此同时,在各类民事和刑事案件, 尤其是经济类案件中,涉及到的以签字笔书写的伪造、 变造文件也越来越多. 在此类案件中, 对签字笔字迹形成时间的检验鉴定意见, 往往成为证明案件事实的重要证据材料. 对于案件中有争议的签字笔书写文件,其主要争点包括文件书写者身份、 字迹的形成时间、 油墨的种类等. 因此, 目前在法庭科学研究领域和司法鉴定工作实务中, 经常需要就可疑文件上的签字笔字迹形成时间进行检验鉴定. 但是,文件字迹书写时间的鉴定却是国际法庭科学界公认的难题之一,[1]一直以来, 可疑文件上字迹形成时间的检验亦是国内外法庭科学界研究的热点和难点问题.
检验字迹的形成时间, 其根本目的是通过对文件字迹的墨水或油墨成分在形成之后随时间流逝的老化规律进行研究, 为确证可疑文件的真实性提供科学依据. 字迹书写于纸张上之后, 染料成分通过吸附、 结膜过程固定在纸张上, 随着字迹形成时间的延长, 油墨成分会发生一系列的氧化、 分解等反应. 形成时间不同的字迹中油墨成分的变化程度不同, 通过分析比较已知形成时间的样本和未知形成时间的检材的油墨成分的变化程度是否相同, 就可以确定二者的形成时间是否相同.
文件检验中的字迹形成时间, 包括绝对形成时间和相对形成时间两种. 绝对形成时间是指文件上字迹形成的具体时间, 即文件字迹是在何年何月何日形成的; 相对形成时间是指检材与样本文件上字迹形成时间的先后顺序. 一般而言, 检验字迹形成的相对时间, 要易于检验字迹形成的绝对时间. 但是, 由于检验字迹形成时间的影响因素较多、 检验所需的主客观条件较苛刻、 操作过程较复杂, 目前还缺乏一种统一、 可靠的标准或方法, 用来检验文件字迹形成时间.
字迹相对形成时间的检验结果, 往往是通过比对未知形成时间检材与已知形成时间样本从而得到的.
针对这一世界性难题, 各国学者先后提出墨水库法、 墨水标记法、 光谱法、 色谱法等多种方法, 并成功实现了从传统方法向现代仪器分析方法的转变. 20 世纪 30 年代以来, 各国法庭科学工作者利用各种现代分析仪器对墨水字迹书写形成时间进行了广泛研究, 取得了一系列成果. 包括运用气相色谱---质谱联用法、[7]液相色谱法、[8]紫外---可见分光光度法、[9-10]薄层色谱法、[11]傅立叶变换红外光谱法、[12-14]拉曼光谱法[15-17]等在内的现代技术手段, 对各种字迹的墨水成分进行分析研究, 判定字迹的形成时间.
对于直到 20 世纪中后期才被发明并风靡世界的签字笔,[18]就其书写字迹的形成时间, 国内外一些实验室利用显微拉曼、 显微红外等光谱技术[15-16,19-21]以及高效液相色谱、[22]质谱[23]等手段进行检验, 并取得相应的研究成果. 我国的夏莉琳等首创使用热重法直接测定可疑文件上墨迹在程序升温燃烧前后的热效应变化, 从而确定字迹形成时间.
目前为止已经发展出来的检验文件字迹形成时间的诸多方法, 就一定对象, 在一定条件下,是成立并有效的. 但是应当指出, 字迹形成时间检验方法尚缺乏系统标准, 具体手段和程序呈现多样化; 成熟、 可靠的字迹形成时间检验方法尚未建立. 而不同的单位, 对于不同的检材和样本,往往采用不同的检验鉴定方法, 乃至形成了不同的 "鉴定流派".
对不同色料的形成时间鉴定, 各个地方均有所长, 所依据的理论都不一样, 更不用说检验方法. 例如, 对于圆珠笔这种过去十分常用, 而目前正逐渐被签字笔取代的书写工具, 对其字迹形成时间的鉴定, 公安部物证鉴定中心书写材料处使用薄层扫描法. 具体要求是, 委托单位必须同时提供检材文件的落款时间附近和怀疑真实书写时间附近的同种类、 相同保管条件的圆珠笔笔迹样本. 比如: 某文件落款 2012 年 5 月, 而怀疑其真实书写时间是在 2013 年 3 月, 就要求提供2012 年 4、 5、 6 月 , 同 时提供 2013 年 2、 3、 4月同种类圆珠笔书写的字迹, 最好是写在相似的纸上, 有着相似的保管条件. 他们采用的方法是剪取少量笔画, 溶解后进行薄层层析, 一般能分出五个以上斑点, 再用薄层扫描仪比较检材与两个时间段样本的斑点分布和大小. 如果检材与2012 年 5 月的样本笔画斑点相近, 就认为检材是2012 年 5 月 书写 ; 如果和 2013 年 3 月 相近 , 就认为是 2013 年 3 月书写. 刑警学院文检教研室采用的方法是弱、 强溶剂提取法. 具体操作是采用打孔针 (直径小于 1mm) 在笔画中提取定量圆珠笔油, 用弱、 强溶剂依次定量提取后进行薄层扫描, 计算弱、 强溶剂提取油墨量的比值, 比值越小, 书写时间越长. 当然也对样本有苛刻的要求(同公安部物证鉴定中心之要求). 司 法部鉴定中心和西南政法大学司法鉴定所采用的方法也是薄层扫描, 但具体方法未见披露.
二、薄层色谱技术的研究发展现状
薄层色谱常用 TLC 表示, 又称薄层层析, 属于固、 液吸附色谱, 是最古老的色谱技术之一,具有简便、 快速、 灵敏等特点. 但由于仪器自动化程度、 分辨率以及重现性等方面的不足, 该技术较长时间停留在定性及半定量的分析水平. 为了充分发挥这一技术的优势, 20 世纪 70 年代以来, 诸多学者和技术人员为薄层色谱的仪器化、高效化和定量化进行了大量的研究和改进工作.
今天, 薄层色谱法是指仪器化高效薄层色谱法. 它已成为公认的、 应用广泛的通用分析方法.
薄层色谱法突出的特点是许多种混合样品可同时分离, 并且唯有 TLC 能够提供图像用以直接观测并传达色谱结果, 色谱的展开结果显而易见, 可以在无须知晓每个斑点色谱的化学性质的情况下,仅通过视觉印象的不同, 对同一混合样品的不同组分, 以及不同种类的样品进行迅速的定性区分;其定量分析速度取决于薄层色谱图的仪器扫描.
因此, 薄层色谱以及薄层扫描技术已经成为分析速度快, 而且成本较为低廉的微量分析技术. 另外, 薄层色谱法对样品预处理要求较低, 对固定相和流动相选择具有较大的自由度, 十分适合于样品中含有不易从分离介质脱附的分析和需要色谱后衍生化处理的样品分析. 目前, 薄层色谱技术已在食品和饮料的成分测定、 草药的质量控制、环境污染监控、 毒品毒物分析、 司法鉴定等领域发挥巨大作用.
(一) 薄层色谱法
将固定相涂布在载板上, 使之形成均匀的薄层; 将待分离的以提取剂为溶剂、 检材物质作为溶 质 的 样 品 溶 液 点 样 在 薄 层 板 上 离 下 沿 约 为10mm 的 位置 ; 将薄层板下沿向下 , 放入盛有深度约 5mm 的展开剂的密闭层析缸中, 进行上行展开. 在色谱展开过程中, 样品的成分受到多种力的作用. 首先是流动相利用毛细作用带着样品穿过固定相. 另外, 还包括样品与固定相之间的相互作用力, 即样品组分在移行过程中发生的偶极、偶极相互作用、 氢键、 范德华力等, 进而使不同组分产生了不同程度的延缓、 分散、 吸附、 离子交换和络合行为, 从而实现混合物中不同组分的分离. 对被展开的色谱谱带 (斑点状或长条状),大多数组分斑点可在日光灯或紫外灯下进行定性检视, 少数组分须经衍生化处理后检视, 同时可以立即采用照相的方法对色谱谱带加以固定, 以防斑点随时间流逝而老化、 褪色. 此外, 还可通过薄层扫描法对组分进行进一步定量分析.
需要注意的是, 在进行薄层展开之前, 最好先进行展开剂蒸汽预饱和. 展开缸内展开剂的蒸汽饱和程度会显着影响样品的色谱行为, 在使用混合展开剂时这一情况尤为突出. 在展开过程中,极性较弱、 沸点较低的溶剂在薄层板边缘容易挥发, 致使薄层板边缘部分展开剂中极性溶剂的比例增大, 使得展开组分的移行偏大, 进而出现同一物质在同一薄层板上中间部分的移行距离比边缘部分的移行距离小, 这种现象被称为 "边缘效应". 若在展开前先将展开槽与薄层板用展开剂蒸汽饱和一段时间后再展开, 边缘效应可大大减轻,利用双槽展开缸尤为便捷.


TLC 是一项离线色谱技术, 所有步骤 (包括点样、 展开、 衍生化、 拍照、 检视、 扫描和计算等) 相对独立, 按照时空分割, 这就使其操作和应用具有很强的灵活性. 在一块薄层板上, 所有的展开样品在完全一致的条件下进行分离; 展开后的色谱谱带保留在薄层板上, 可进行多次分析计算, 不必对同种样品重复分离.
(二) 薄层扫描法
这是一种以薄层色谱法为基础发展起来的TLC 组分原位分析方法及薄层色谱的记录 方法 .它利用薄层扫描仪, 以一定波长的光照射展开后的薄层板上的斑点, 测定其对光的吸收或所发出的荧光, 进行定量分析的方法. 其具体原理与双波长分光光度计相似. 从光源发出的光, 通过两个单色器分光后, 成为不同波长的两束光 λ1和λ2. 斩光器使它们交替地照射到薄层板上 , 经透射或反射后分别由光电倍增管接受, 再输出电信号, 由对数放大器变换成吸光度, 记录下的信号是 λ1和 λ2两波长吸光度之差. 通常是选择斑点中化合物的吸收峰波长作为特定波长, 选择化合物吸收光谱的基线部分作为参比波长.
用一定波长的光束对薄层板上的斑点谱带进行扫描, 记录其吸光度随展开距离的变化, 得到薄层色谱扫描曲线, 曲线上每一个吸收峰代表薄层谱带上的一个斑点. 吸收峰高或峰面积与斑点组分的物质含量之间有一定的关系, 比较对照品与样品的峰高或者峰面积, 可得出样品待测组分的相对含量.

三、利用薄层色谱技术检验签字笔字迹相对形成时间的方法.
如前所述, 尽管目前利用各种现代技术和仪器检测签字笔字迹形成时间的方法很多, 但相较于薄层色谱技术, 很多时候仅仅为了得到一个简单的检验结果而投入较大的时间、 财力和物力,去使用其他昂贵的现代仪器分析技术进行书写时间鉴定, 是条件不允许或者不够经济、 没有必要的. 另一方面, 混合物通过薄层色谱展开后得到的彩色斑点和条带在混合物的成分分析中显得十分有用, 只需一张图像, 作为直观的扩展信息,并且可以方便地借助薄层扫描技术得到更多定量数据. 薄层色谱不但是一种应用广泛的分析手段,并且可以以很小的投入得到很好的效果, 同时也可以作为其他技术的补充. 因此, 介绍一种利用薄层色谱技术检验签字笔字迹相对形成时间的方法.
本方法的基本思路是, 利用薄层色谱法和薄层扫描法, 对待检验的签字笔字迹检材和样本进行油墨提取、 有机溶剂溶解, 再将油墨样品溶液薄层展开, 并对展开后的谱带进行薄层扫描, 进而获取薄层色谱扫描曲线和谱带峰面积数据, 最终通过分别计算检材与样本的峰值比, 并对其进行对应比较, 确定检材的相对书写时间.
应当指出的是, 本方法适用于油墨能够溶于有机溶剂 (包括提取剂和展开剂, 例如笔者使用的是, 提取剂---蒸馏水: 无水乙醇=1∶1, 另外加入几滴冰醋酸. 展开剂---正丁醇: 无水乙醇:
蒸馏水: 冰醋酸=6∶2∶2∶1) 的签字笔或圆珠笔字迹, 但不适用于以极细碳颗粒为主要着色成分的油墨形成的字迹. 因其着色成分碳颗粒无法被一般有机溶剂溶解, 故无法进行薄层展开. 因此,在薄层实验开始之前, 务必要通过预实验或其他方法确定字迹油墨能否溶于有机溶剂.
下面举例说明本方法中峰值比的概念、 计算与比较.
(一) 峰值比的概念、 计算

将图 4 所示的展开后的薄层板放入图 3 所示的薄层扫描仪的扫描平台. 手动调节平台, 其坐标在电脑屏幕显示, 通过键盘和鼠标输入合适的参数, 选择合适的扫描波段, 关闭平台舱门, 由软件自动控制扫描过程, 沿薄层板上各个点样展开的谱带 (纵列) 进行扫描, 获得薄层扫描图像及数据.
取其中一条谱带 (Track 1) 为例, 获得其薄层扫描图像及数据分别如图 5、 图 6 所示.
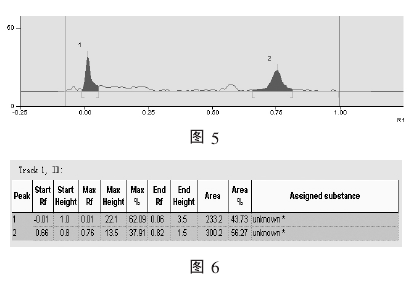
Track 1 上有两个斑点, 对应图 5 上的两个深色吸收峰 (Peak 1 和 Peak 2). 峰面积的大小反映了斑点中色料成分含量的多少.
阅读图 6 的数据可知, 吸收峰一 (Peak 1)的峰面积 (Area 1) 为 233.2, 吸收峰二 (Peak 2)的峰面积 (Area 2) 为 300.2.
为排除取样量、 书写力度、 笔道粗细及出墨水量等随机性因素的干扰, 需要以各种染料成分的相对含量的变化作为考察指标. 所以我们选择峰值比代替峰面积, 作为衡量书写时间长短的尺度.所谓峰值比, 即除面积最大的吸收峰之外的所有吸收峰的峰面积与面积最大的吸收峰的峰面积的比值. 设某种样品经薄层分离和扫描后, 获得了 (n+1) 个吸收峰 (n∈N+), 其中面积最大的吸收峰的峰面积为 S0, 其他吸收峰的峰面积为 S1,S2, S3……Sn, 则有 n 个峰值比, 分别为 S1/ S0, S2/ S0, S3/ S0……Sn/ S0.
在图 5 和图 6 的例子中, 共有两个吸收峰,n=1, S0=300.2, S1=233.2, 所以可计算出一个峰值比: S1/ S0=233.2÷300.2≈0.7768=77.68%.
(二) 峰值比的比较及字迹相对形成时间的判断
对于同种油墨 (对于不同种油墨的鉴定意见,一般即可据以认定检材文件为篡改或伪造文件,而无需进一步确定字迹形成时间) 在不同时间形成的检材与样本字迹, 经过上述一系列检测和计算, 获取每一条谱带 (即每种样品溶液) 的峰值比后, 需要分别对其进行数据的平行比对和分析.
例如, 一般而言, 同种油墨在不同时间形成的字迹, 虽然相应吸收峰的峰面积和峰值比发生变化,但吸收峰的个数和位置应保持不变. 对于检材,得出了 n 个峰值比, 分别为 S1/ S0, S2/ S0, S3/ S0……Sn/ S0; 对于样本, 亦得出了 n 个峰值比, 分别为 S1′ / S0′, S2′ / S0′, S3′ / S0′… …Sn′ / S0′ . 尔 后 ,分别比较 S1/ S0与 S1′ / S0′, S2/ S0与 S2′ / S0′… …Sn/S0与 Sn′ / S0′, 并计算 n 个峰值比的变化量, 即|S1/S0-S1′ / S0′|, |S2/ S0-S2′ / S0′|……|Sn/ S0-Sn′ / S0′|.
若 n 个峰值比的变化量均较小, 则说明检材与样本字迹在形成时间上相近; 若 n 个峰值比的变化量均较大, 则说明检材与样本字迹在形成时间上相差较大. 在理想状态下, 同时书写的检材和样本字迹, 其峰值比变化量应为 0; 但是实验数据不可避免地存在着误差, 在实际情况中, 即便为同时书写的检材和样本字迹, 其峰值比变化量也很难为 0, 而应存在一个正的误差量. 因而,如何确定据以判断变化量 "较小"、 "较大" 的临界值, 是本方法的一个关键点和难点. 具体而言,应当根据一定的技术和设备条件, 通过实验确定该临界值, 做到具体问题具体分析. 笔者认为,在确保一定实验精度下, 5%是一个供参考的临界值.
最后需要指出的是, 本方法检验结果的客观准确性受到两个因素的影响和制约.
第一, 对于同种油墨在不同时间形成的检材与样本字迹, 如果两者形成的时间差很小, 例如数小时或数天, 则峰值比的相对变化幅度一般不够明显; 如果两者均已形成很长时间, 例如数年甚至十几年, 由于其主要成分的氧化进程基本完成,[27]峰值比趋于稳定, 其峰值比的相对变化幅度亦不十分明显. 由此可以推论, 当峰值比变化量较小时, 既不能断定检材与样本字迹一定是连续书写形成的; 亦不能排除检材与样本字迹均已形成很长时间, 难以确定相对形成时间的可能.
出现这种情况, 鉴定意见中应当如实说明, 并应结合其他证据材料进一步判断.
第二, 文件的保管环境和文件人为老化手段会影响实验结果的可靠性. 具体来说, 同一份文件, 在密闭缺氧的环境中保存和在风吹日晒的环境中保存同样的时间, 其老化程度反映在峰值比的变化量上是完全不同的. 又如, 不法行为人可以通过紫外线照射文件等方法, 加速文件的老化速率. 这些可能性, 无论在在检验鉴定过程中,还是在证据材料的审查判断中, 都是应当考虑在内的. 一言以蔽之, 就是要做到具体问题具体分析.
参考文献:
[1] 贾玉文, 邹明理等. 中国刑事科学技术大全---文件检验[M].北京:中国人民公安大学出版社,2002:1176-1228.
[2]王世全,贾玉文.文件制成时间鉴别方 法 的评价[A].全国文件制成时间鉴定研讨会,2007.
[3]谢鹏,王英英等.透析法测定文件字迹 书写时间的研究[J].广东公安科技,2009,(2):8.
[4]Aginsky VN. Some New Ideas for Dating Ball-point Inks-a Feasibility Study [J].Journal of ForensicSciences,1993, (5):1134 -1150.
[5]Aginsky VN. Current Methods Dating Ink -which is the best? The 49th Annual Meeting of theAmerican Academy of Forensic Sciences,1997:17- 22.
[6]Andrasko J. HPLC Analysis of Ballpoint PenInks Stored at Different Light Conditions [J]. Journal ofForensic Sciences,2001,(1): 20-31.
[7]Weyermann C, Kirsch D, Vera C C, et al. AGC/ MS Study of the Drying of Ballpoint Pen Ink onPaper [J]. Forensic Science International, 2007, (2):119-127.
[8]史晓凡,李心倩,许英健等.高效液相色谱法鉴定蓝色圆珠笔油墨字迹的书写时间[J].光谱学与光谱分析,2006,(9):1765-1768.

随着建筑业多元化发展,对工程建设实施阶段的造价控制提出更高要求,为更好适应建设领域当前发展趋势,工程造价鉴定部门需结合工程实际情况,制定出适合该工程鉴定完善的工程造价鉴定措施与方法,充分发挥出工程造价鉴定工作,实现工程综合效益最大化起到积极作用。...

一、痕迹检验技术的内容以及重要意义(一)痕迹检验技术的概述痕迹检验技术的主要工作原理是在刑侦工作过程中,通过痕迹检验的相关的理论以及方式进一步提升证据的质量,同时收集以及检验犯罪现场中遗留的各种痕迹。这样做既能够有效的推断出犯罪变化的规...
痕迹检验技术就是运用痕迹检验学的相关方法和理论,对案件中的各种物证痕迹进行检验,从而确定痕迹与案件、人物关系的一种刑事破案技术。通过长期的实践经验可以看出:痕迹检验技术逐步成为案件侦破不可或缺的重要手段,而痕迹检验工作的细致与否也关系着案...

一、引言二、锡纸开锁工具组成简介技术开锁因其隐蔽性和便捷性的特点,成为犯罪分子侵入防盗门实施入室盗窃的主要手段.其中,锡纸技术开锁方式工具轻巧、上手简易、开启时间短暂,在近两年技术开锁案中的使用比例越来越大,对社会治安构成了严重危害.在此,笔...
随着系统论的思想被引入笔迹检验,对笔迹多层次属性的进一步研究也势在必行。在笔迹检验中,以笔迹多层次属性为基础,才能正确认识笔迹,分析笔迹,系统开展笔迹检验。因此,研究笔迹的多层次,对于丰富笔迹检验理论和检验技术都有着极其重要的意义。一、笔...
指纹上的汗孔、纹线边缘、细点线、疤痕等特征是更加细节的指纹三级特征。对于低质量指纹、残缺指纹等二级特征数量少的指纹识别具有一定的鉴定价值。不论是人工指纹鉴定实践中,还是指纹自动识别系统中,利用一级、二级细节特征外,还希望能利用这些三级特征。因...
一、引言随着科学技术的飞速发展,犯罪分子的作案手段也日趋科技化,这无疑给公安机关侦查人员的案件侦破工作带来了更大的困难。因此,引入先进的科学技术手段来帮助侦查案件是当代刑事侦查工作中十分重要的环节。手印痕迹检验技术在刑侦工作中扮演着重要的...
一、引言各类窗、门,各种器皿、餐具、杯、盘、碟等玻璃制品,是人们日常生活中不可缺少的生活用品。在很多现场中,案犯都是通过对玻璃的破坏而开启门窗进入现...
毒品危害是广泛深远的,其损害人类健康,破坏家庭和睦,诱发违法犯罪,破坏社会稳定。要打击毒品犯罪,首先就需要对毒品进行准确判定,包括毒品种类及纯度含量等。目前市面上的毒品种类繁多、添加成分多样,这增加了毒品种类鉴别和纯度分析工作的难度[1-3]....
盗窃案件现场痕迹包括作案人员的DNA、足迹、手印以及能帮助破获案件的有效痕迹等,是破获案件的重要证据与线索,因此,有必要加强盗窃案件现场痕迹寻找方法的研究,以提高盗窃案件破获率,营造安全的社会治安与环境。一、案例分析2015年04月06日22时30分至...