分析化学论文

摘 要: 建立了一种芯片液相色谱-高分辨质谱测定3种微囊藻毒素 (MC-RR、MC-YR和MC-LR) 的分析方法.在最优条件下, 3种藻毒素在16 min内实现基线分离, 并选择电喷雾源正离子模式进行质谱测定.方法在2.5~2 000 ng/m L范围内线性良好, 线性相关系数大于0.996 5, 检测限介于0.5~2.0 ng/m L之间.鱼肉和虾肉样品经固相萃取法净化处理后未测得上述毒素残留, 加标回收率为82.8%~120.3%.方法具有样品用量少、定性快速准确、灵敏度高等优点, 适合用于水产品等生物样品中藻毒素的痕量监测.
关键词: 微囊藻毒素; 芯片液相色谱; 质谱; 水产品;
Abstract: A method for the simultaneous analysis of 3 microcystins ( MC-RR, MC-YR and MC-LR) was set up by the nano-flow chip liquid chromatography-high resolution mass spectrometry. Under optimal conditions, the mentioned microcystins were baseline separated in 16 min with an ESI source in the positive ion mode. Good linear range in 2.5 ~ 2 000 ng/m L was obtained, with the correlation coefficient larger than 0.996 5 and the limit of detection between 0.5 ~ 2.0 ng/m L.Solid phase extraction method was used for the extraction and clean-up of fish and shrimp samples. No residue of the three microcystins was detected and the spiked recoveries were between 82. 8% ~ 120. 3%. The developed method is sensitive, rapid and exact with low sample consumption, so it is reliable for the trace determination of microcystins in biological samples including aquatic products.
Keyword: microcystins; nano-flow chip liquid chromatography; mass spectrometry; aquatic product;
蓝藻水华是常见的淡水水华现象, 产生的毒素以微囊藻毒素 (MCs) 最普遍且危害最大[1,2].大量藻毒素的存在不仅影响生态环境, 还会通过食物链富集和传递, 影响淡水养殖业并威胁人类健康.MCs是一类由7个氨基酸残基组成的环状多肽化合物目前已发现了几十种该类毒素, 其中MC-RR、MC-LR和MC-YR最具有代表性, 其测定技术包括生物学法、免疫检测法和化学分析法等[3].化学分析法中, HPLC是推荐的MCs测定方法.痕量毒素通常需要先浓缩富集再经HPLC-UV法直接测定[4,5].此法结合质谱检测器, 方法的灵敏度和定性准确性明显提高, 但较大的进样量和较低的柱效在常规HPLC-MS或HPLC-MS/MS中略显不足[6,7].因此, 开发一种高效、样品消耗少的MCs检测方法仍具有实际意义.
芯片色谱技术把传统色谱柱浓缩成约数厘米长、数毫米厚的芯片, 再通过激光镭射技术在色谱芯片上刻出分析通道, 并填充C18、C8等固定相.该微型化装置可明显降低流速、减少柱体积, 从而提高方法灵敏度[8,9].目前芯片液相色谱-质谱 (nano-flow chip liquid chromatography-high resolution mass, nano-flow chip LC-MS) 技术已成功应用于蛋白质、肽类等生物样品分析[10,11,12], 但较少用于小分子化合物领域[13,14,15], 在MCs定量监测中也鲜见报道.

本文将芯片液相色谱和高分辨质谱技术相结合, 建立了同时检测MC-RR、MC-LR和MC-YR的nano-flow chip LC-MS新体系.方法快速可靠, 仅pg级的进样量也获得了较高的灵敏度.
1、 试验部分
1.1、 仪器与试剂
Nano-flow chip LC-MS联用系统 (Agilent, USA) 包括纳流液相色谱 (1200 Nano HPLC) 、接口 (Chip Cube) 和四级杆-飞行时间质谱仪 (G6520Q-TOF MS) 3部分.芯片为Agilent G4240-65005, 包括富集柱 (体积40 nL) 和分析柱 (75μm×43 mm) , 二者填料均为Zorbax SB-C18, 5μm (如图1所示) .
图1 MCs的化学结构 (a) 和芯片示意图 (b)
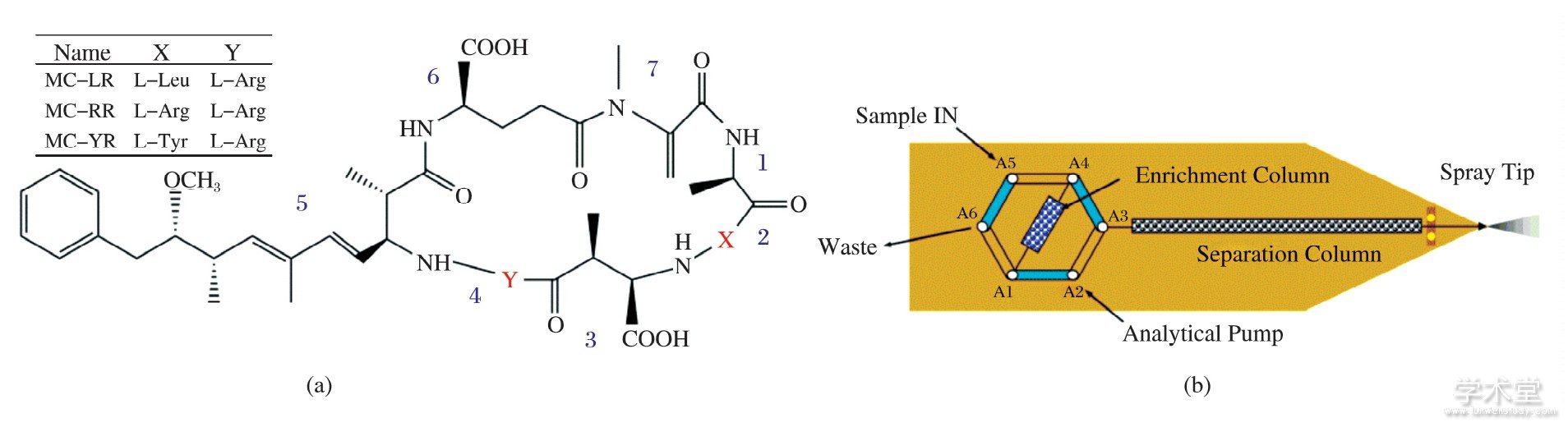
Fig.1 Chemical structure of MCs (a) and schematic diagram of chip (b)
标准品MC-RR、MC-LR和MC-YR (北京伊普瑞斯公司, 结构式如图1所示) 使用甲醇∶水 (体积比为2∶8) 溶解, 配制成100.0μg/m L储备液, 试验前用二次水稀释至所需浓度.乙腈为色谱纯 (美国Sigma-aldrich公司) ;醋酸铵为分析纯 (天津市福晨化学试剂厂) ;甲酸 (FA) 为色谱纯 (上海迪马科技有限公司) .Bond Elut-C18固相萃取柱 (Agilent, USA) .试验用水为二次蒸馏水, 取自Milli-Q超纯水系统.所有溶液使用前均用0.22μm滤膜过滤.
1.2、 芯片液相色谱-质谱条件
毛细管泵流速2μL/min, 泵冲洗选择乙腈-水溶液, 10%乙腈等度洗脱.纳流泵流速600 n L/min, 乙腈 (B相) -水溶液 (A相, 含0.15%FA) .0~20 min, 10%~60%B相梯度洗脱.进样体积lμL, 进样冲洗体积为4μL.
质谱选择正离子模式;m/z 400-1200, 干燥气流量3 L/min, 雾化气温度315℃, Fragmentor 175 V, 毛细管电压调节在1 750 V使喷雾良好.3种MCs均测得[M+H]+、[M+2H]2+信号, 试验以[M+2H]2+作为定量离子 (如表1所列) .
1.3、 样品处理
明虾和草鱼购于福州当地超市.采用固相萃取法 (SPE) 提取净化样品, 过程在文献[7]基础上稍加改进.准确称取鱼肉或虾肉组织1.0 g, 加入5.0 m L 5%醋酸铵溶液, 匀浆搅碎, 超声20 min, 浸提4 h.过滤, 残渣用2.5 m L 5%醋酸铵溶液重复提取两次, 合并滤液.分别用甲醇、水活化处理C18小柱后, 让滤液以一定速度上样富集藻毒素.待样品全部吸附, 以5.0 m L 10%甲醇-水溶液冲洗C18小柱, 5.0 m L甲醇洗脱目标物, 洗脱液氮吹近干, 用二次水定容至1.0 m L, 15 000 r/min离心10 min, 取上清液进行色谱、质谱分析.
表1 3种MCs的保留时间和特征离子
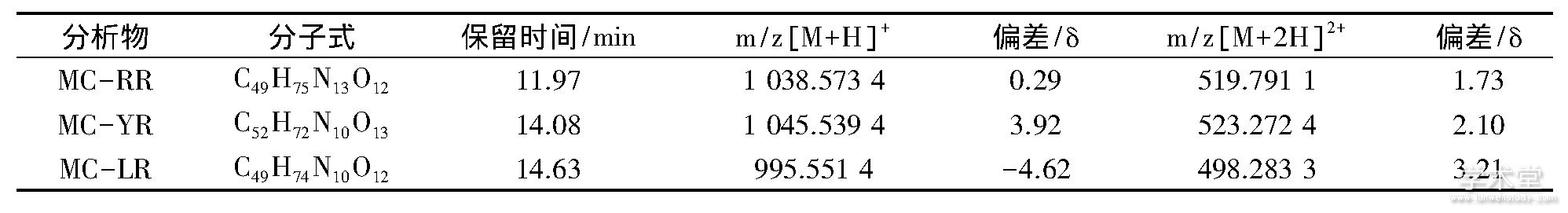
2、 结果与讨论
2.1、 芯片液相色谱分离优化
芯片液相色谱高度集成了芯片色谱柱分离和预富集、纳流喷雾电离等技术, 高度智能化、微型化的同时也具有较好的色谱分离性能.
2.1.1、 样品溶剂和上样体积的选择
在芯片中, 样品先预富集在富集柱上 (柱体积仅40 nL) , 达到设定的上样体积时, 六通阀切换使样品在纳流泵的冲洗下进入分析柱 (图1b) .若稀释样品的溶剂洗脱强度较大, 则可能导致在预富集柱中的样品部分排入废液或后续分离时峰展宽.试验考察了5%乙腈-水、2%乙腈-水、水作为样品溶剂时, 样品的分离效果.结果发现, 即使添加少量乙腈, 3种分析物峰形都略微展宽, 最终选择使用水作为样品溶剂.
选择合适的上样体积, 对样品的测定非常重要体积太小, 不能保证将所有目标物运送至富集柱;若体积太大, 可能会导致弱保留化合物穿透富集柱鉴于上样体积一般为针座毛细管体积、自动进样器到Chip Cube之间毛细管体积、在线过滤器体积之和的2~6倍, 试验选择4μL为上样体积.
2.1.2、 流动相选择
对于芯片色谱而言, 流动相对色谱分离的影响甚大.芯片的耐受压力约为15 MPa, 若使用甲醇-水体系, 较高的压力可能超过芯片的承受范围.使用乙腈-水体系, 系统压力更低, 而且乙腈具有较强的洗脱能力, 能够获得更好的峰形.因此, 选择乙腈-水作为流动相.
MCs结构中含有多种氨基酸, 其极性会随pH值改变而变化.试验分别考察了甲酸体积分数为0.05%~0.15%时, 目标物的分离情况 (如图2所示) 由图2可见, 仅使用乙腈-水二元体系时, MC-YR和MC-LR基本重合.随着甲酸体积浓度增大, 3种物质的分离度增大, 响应增强.这是因为随着甲酸浓度的增加, 促进了MCs的质子化过程和极性差异情况, 从而明显改善了各组分在芯片上的保留行为.因此, 最终选取体积分数为0.15%的甲酸作为水相添加剂.
图2 不同流动相对分离的影响
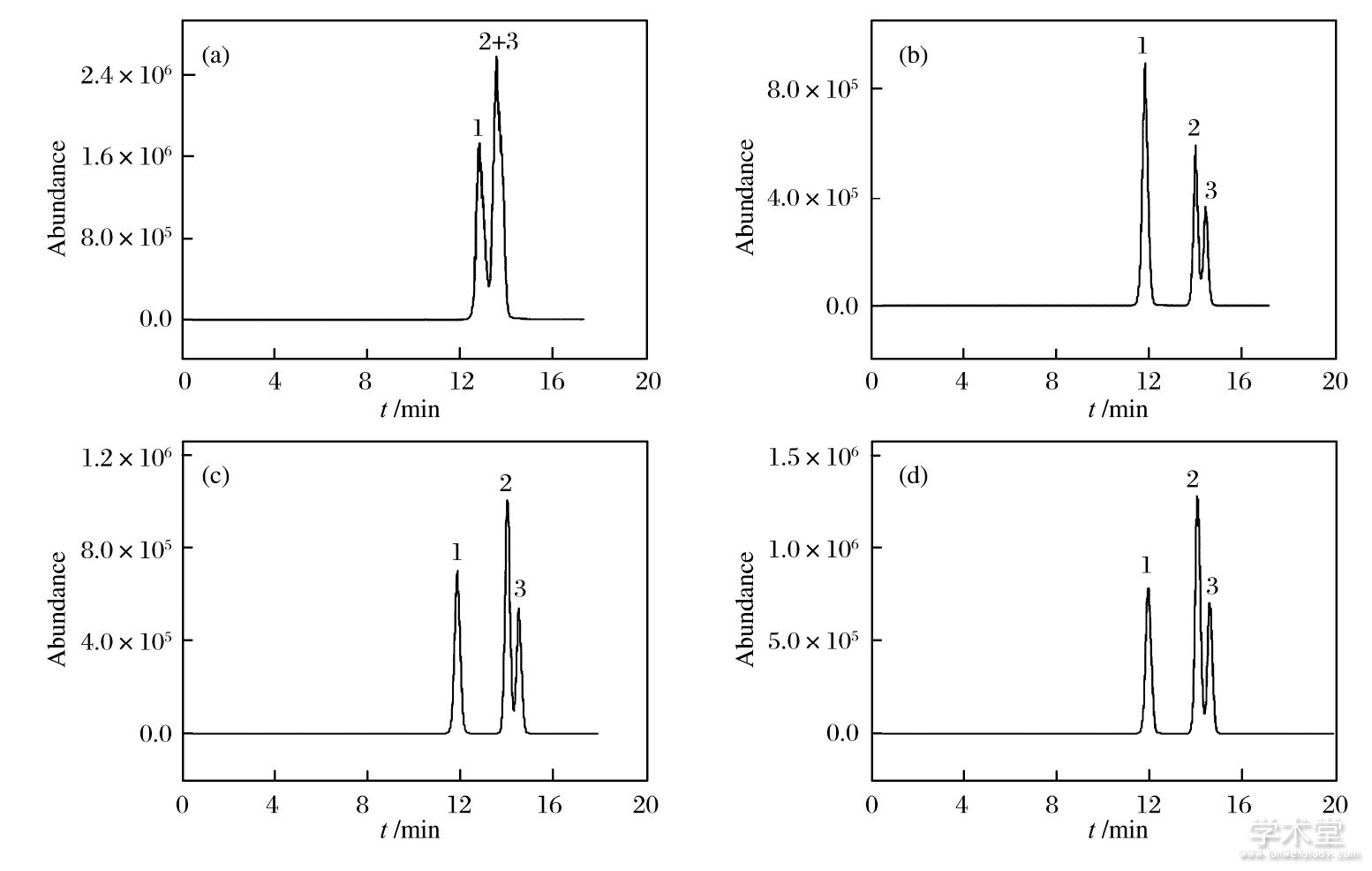
Fig.2 Effects of different mobile phase on separation
(a) 乙腈-H2O, (b) 乙腈-0.05%FA, (c) 乙腈-0.10%FA, (d) 乙腈-0.15%FA毛细管泵流速2μL/min, 10%乙腈等度洗脱;纳流泵流速600 n L/min, 0~20 min, 10-60%B相梯度洗脱进样体积lμL, 进样冲洗体积为4μL (1) MC-RR (0.5μg/m L) , (2) MC-YR (2.0μg/m L) , (3) MC-LR (1.0μg/m L)
梯度洗脱有助于进一步改善分离效果.尝试了多种梯度洗脱程序 (如图3所示) , 发现增大泵流速或有机相比例, 可使保留时间缩短.以20%乙腈为初始有机相冲洗色谱柱时, 尽管3种待测物在14 min内完成分离, 但柱效和灵敏度都明显下降.兼顾分离效率和方法灵敏度, 选择0~20 min、10%~60%B作为后续分析条件.
图3 不同梯度洗脱条件对分离的影响
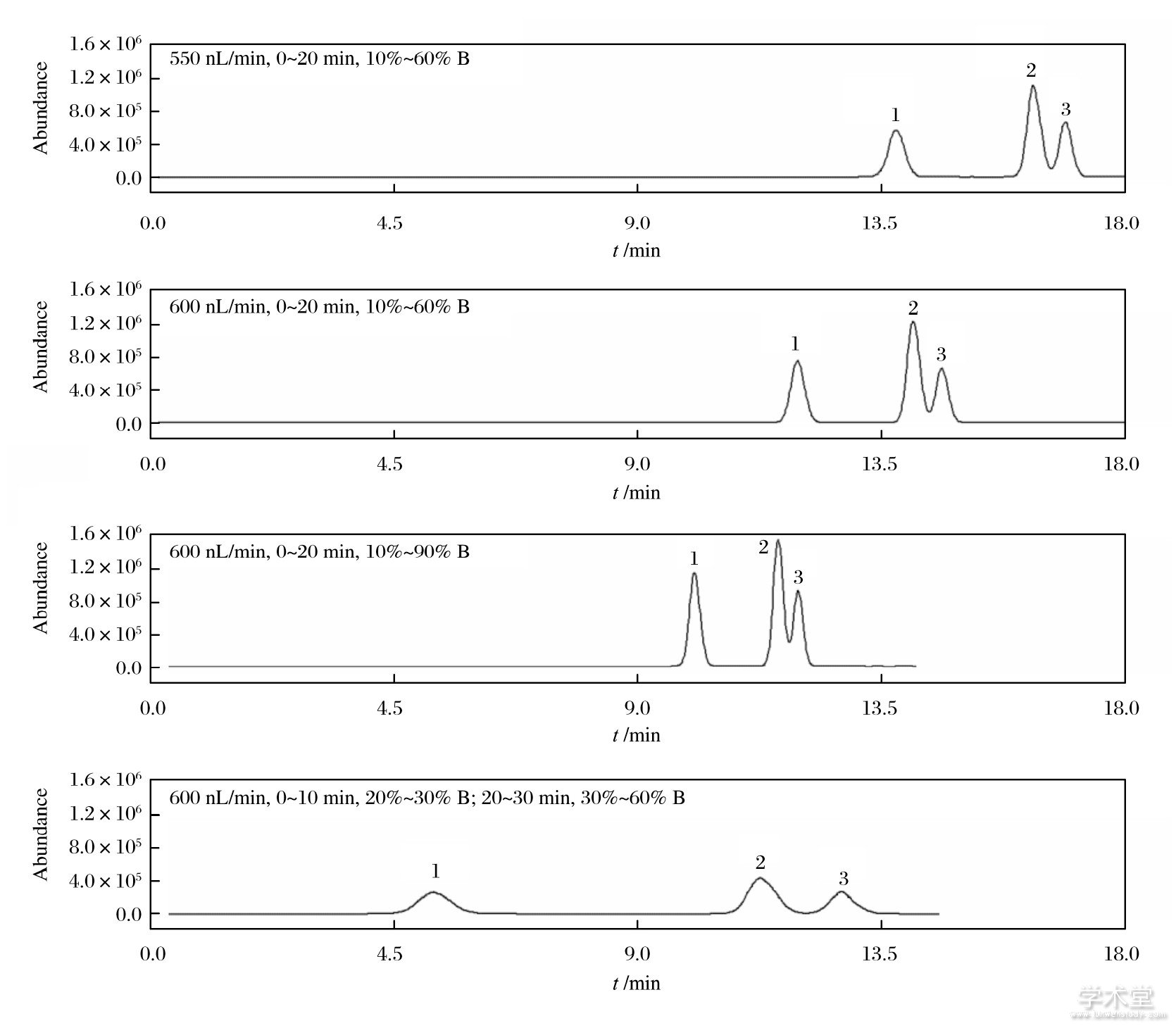
Fig.3 Effects of different gradient elutions on separation
乙腈-0.15%FA;毛细管泵流速2μL/min, 10%乙腈等度洗脱;进样体积1μL, 进样冲洗体积为4μL (1) MC-RR (0.5μg/m L) , (2) MC-YR (2.0μg/m L) , (3) MC-LR (1.0μg/m L)
2.2、 质谱检测
在芯片液相色谱-质谱中, 流动相组成、芯片喷针位置、芯片差异等因素都会影响纳流喷雾效果.在色谱分离方面, 已证实流动相中添加少量FA有利于改善喷雾情况.试验继续考察芯片因素对分离检测的影响, 结果显示, 微调芯片喷针位置, 目标物的响应信号会随之改变.故所有试验应确保在同一个芯片上进行, 且喷针位置一经确定后不再改变.根据喷雾状态优化毛细管电压 (1 500~2 000 V) , 最终确定为1 750 V, 在此条件下方法灵敏度更高.
MCs结构中含有氨基、羧基和酰胺基, 理论上对质谱的正、负两种离子检测模式都有响应.试验优化了上述两种检测模式下的物质信号情况, 也证实了含氮杂原子的存在, 使得正离子检测模式下目标物的响应峰面积更高.3种MCs均测得[M+H]+、[M+2H]2+信号, 选择响应较强的[M+2H]2+作为定量离子 (如图4所示) .在高分辨质谱中, 所有目标物的质量偏差更小, 定性准确性更强.
图4 正离子检测模式下3种MCs的全扫描质谱图
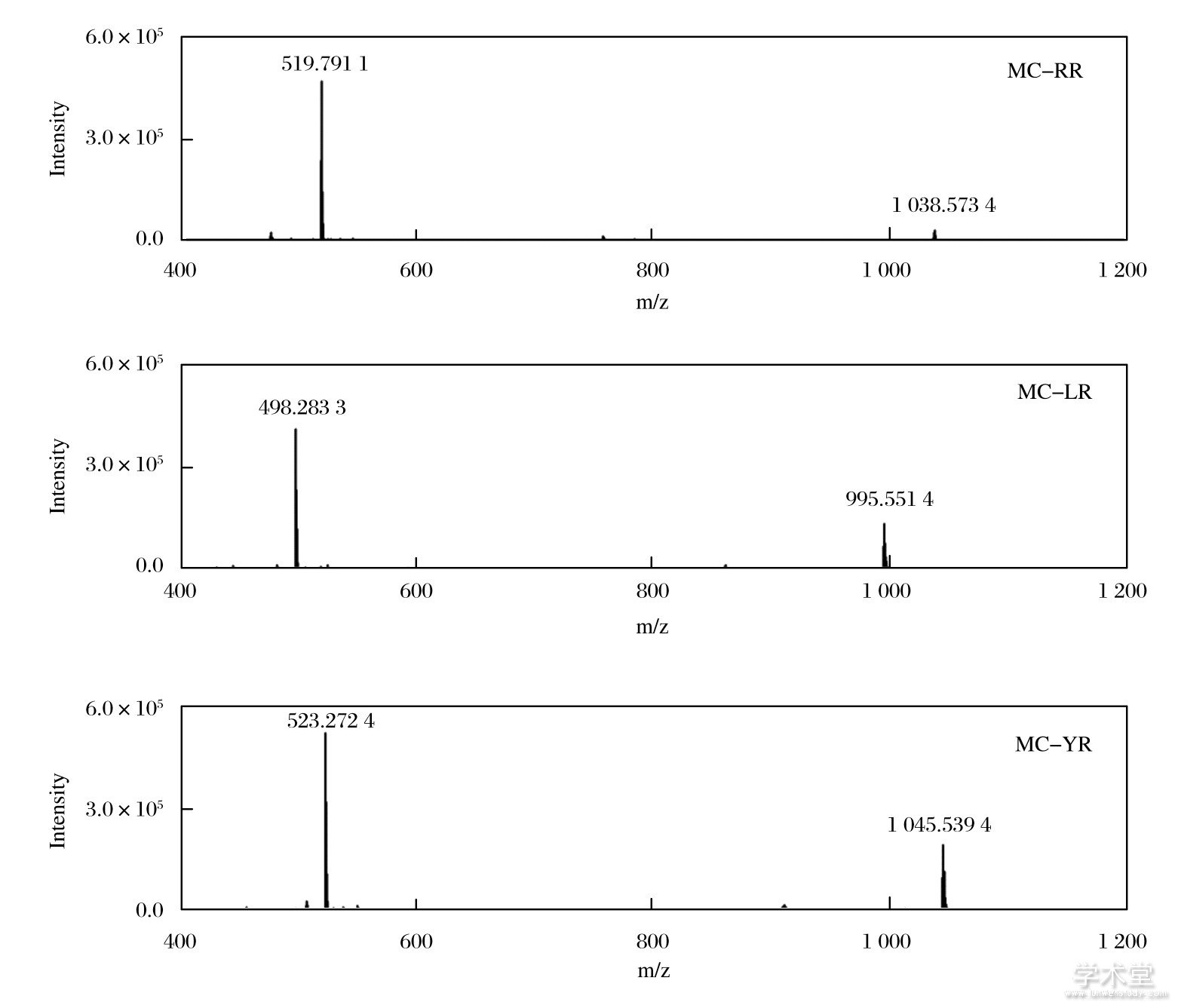
Fig.4 Mass spectra of three MCs under full scan with positive mode
2.3 、定量方法建立
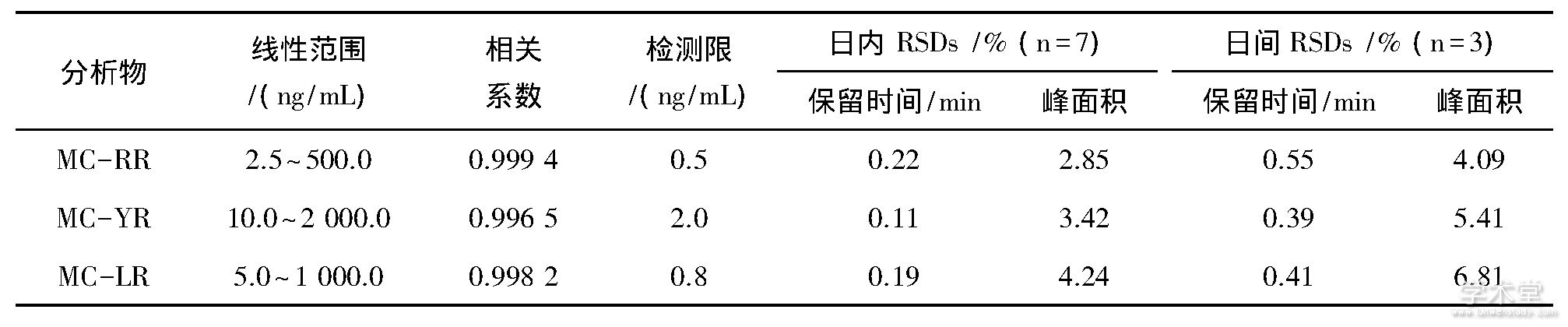
配制一系列不同浓度的MCs标准混合溶液.选择[M+2H]2+的峰面积定量分析, 绘制标准工作曲线.3种MCs在2.5~2 000 ng/m L范围内线性良好, 相关系数大于0.996 5, 检测限 (LOD) 最低为0.5 ng/m L (如表2所列) .目前, 我国对水产品中MCs残留未有明确限量, 但已规定在饮用水中MC-LR限量值为1.0 ng/m L[16].该方法MC-LR的检测限为0.8 ng/m L, 因此能满足痕量MCs日常监测的要求.
考察方法的重现性.日间精密度以当天7次重复进样, 日间精密度以连续3天进样.结果表明, MCs的保留时间稳定, 峰面积RSDs小于6.81%, 方法重现性良好.
表2 3种MCs的分析参数
2.4、 水产品样品分析
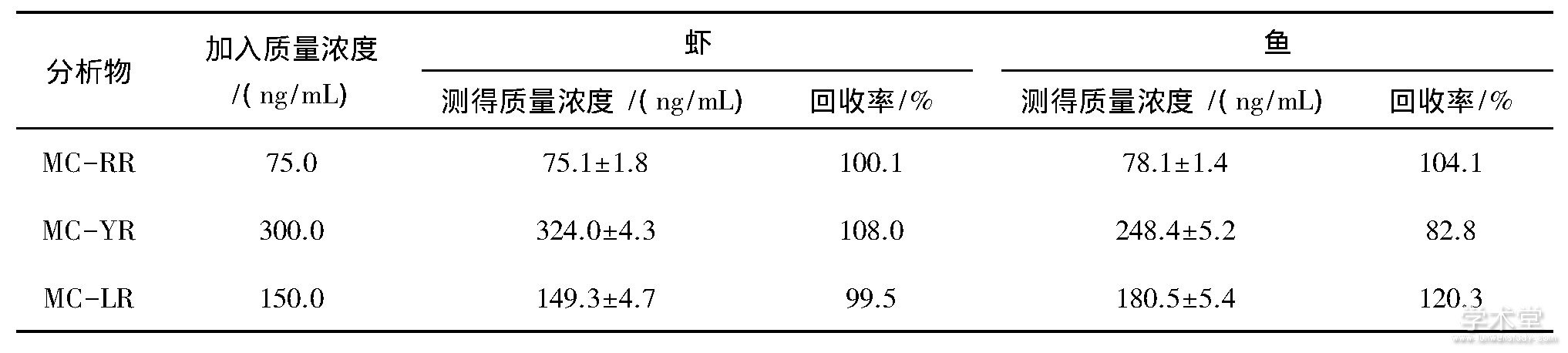
分别对鱼肉和虾肉按照1.3节方法进行处理.样品组织经醋酸铵提取、C18净化富集进行质谱分析.对照目标物保留时间、精确质荷比等信息, 表明在实际样品中未检出MCs.在最优条件下, 进行加标回收率试验 (如表3所列) .所有样品的回收率大于82.8%~120.3%, 能够满足分析方法的要求.
表3 水产品的加标回收率
2.5、 方法对比
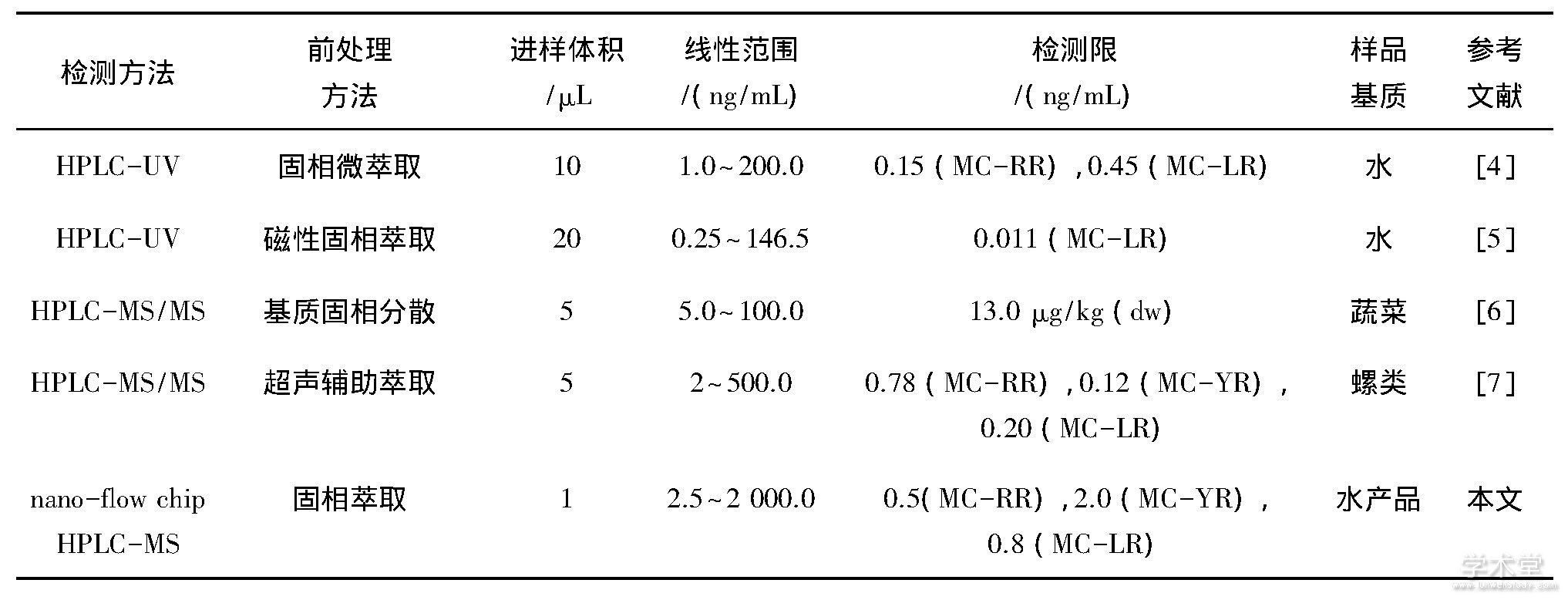
将该方法与已报道的HPLC有关方法相比较 (如表4所列) .由表4可见, 在进样体积方面, 本法的进样体积远低于其他方法, 这对于某些昂贵或稀少样品特别重要, 并符合绿色化学的要求.其次, 结合经典的SPE预处理方法, 本试验的灵敏度可达pg级, 线性范围更广.常规的HPLC-UV法通常需要结合一些新兴样品前处理技术 (例如固相微萃取和磁性固相萃取) 和制备新型吸附材料, 才能获得和本方法接近的灵敏度.此外, 串联质谱技术具有选择离子、提取离子等功能, 可适用于复杂样品分析.综上所述, 本法集成样品预富集、分离、喷雾于一块芯片上, 操作简单、快速, 并具有样品消耗量少、检测限低等突出优势.
表4 本法与HPLC相关方法比较
3 、结论
试验利用nano-flow chip LC-MS技术, 建立了同时分析3种微囊藻毒素的方法.方法检测限在0.5~2.0 ng/m L范围内.采用固相萃取法纯化2种水产品, 均没有测得藻毒素残留.方法简单、灵敏、环境友好, 可推广至其他复杂基质中MCs的快速准确分析.
参考文献
[1] 赵漫, 李冰, 马燕天, 等.应用环境微生物治理淡水湖泊微囊藻毒素污染的研究进展[J].微生物学通报, 2018, 45 (4) :893-899.[ZHAO Man, LI Bing, MAYan-Tian, et al.Advances in the application of environmental microbes to control microcystins in freshwater lake[J].Microbiology China, 2018, 45 (4) :893-899.]
[2]Pham T L, Utsumi M.An overview of the accumulation of microcystins in aquatic ecosystems[J].J Environ Manage, 2018, 213:520-529.
[3] 钟力, 徐文琦, 胡静文, 等.微囊藻毒素的危害及其分析方法进展[J].分析化学进展, 2018, 8 (3) :91-102.[ZHONG Li, XU Wen-qi, HU Jing-wen, et al.The harmful effects of microcystins and the analytical methods[J].Advances in Analytical Chemistry, 2018, 8 (3) :91-102.]
[4] 吴伟文, 杨左军, 顾浩飞, 等.固相微萃取/高效液相色谱法测定水中的微囊藻毒素[J].分析测试学报, 2007, 26 (4) :545-547.[WU Wei-wen, YANG Zuojun, GU Hao-fei, et al.Determination of trace microcystin in water by solid phase microextraction coupled with high performance liquid chromatography[J].Journal of Instrumental Analysis, 2007, 26 (4) :545-547.]
[5] 郭亭秀, 娄大伟, 连丽丽, 等.Fe3O4@Ni Si O3对水中痕量微囊藻毒素的萃取[J].分析测试学报, 2016, 35 (9) :1137-1141.[GUO Ting-xiu, LOU Da-wei, LIAN Li-li, et al.Extraction of trace mierocystins in water using Fe3O4@Ni Si O3[J].Journal of Instrumental Analysis, 2016, 35 (9) :1137-1141.]
[6]Qian Z Y, Li Z G, Ma J, et al.Analysis of trace microcystins in vegetables using matrix solid-phase dispersion followed by high performance liquid chromatography triple-quadrupole mass spectrometry detection[J].Talanta, 2017, 173:101-106.
[7]Li S Y, Cui Y W, Wang Y, et al.A shotgun method for high throughput screening microcystins in margarya melanioides on a triple quadrupole tandem mass spectrometry[J].Food Chem, 2018, 269:89-95.
[8]Haghighi F, Talebpour Z, Nezhad A S.Towards fully integrated liquid chromatography on a chip:evolution and evaluation[J].Tr AC Trends Anal Chem, 2018, 105:302-337.
[9]Yin H, Killeen K.The fundamental aspects and applications of agilent HPLC-Chip[J].J Sep Sci, 2007, 30 (10) :1427-1434.
[10]Li X J, Fekete A, Englmann M, et al.At-line coupling of UPLC to chip-electrospray-FTICR-MS[J].Anal Bioanal Chem, 2007, 389 (5) :1439-1446.
[11]Zhang Y, Li Y Y, Qiu F, et al.Comparative analysis of the human urinary proteome by 1D SDS-PAGE and chip-HPLC-MS/MS identification of the AACTputative urinary biomarker[J].J Chromatogr B, 2010, 878 (32) , 3395-3401.
[12]Kozlik P, Sanda M, Goldman R.Nano reversed phase versus nano hydrophilic interaction liquid chromatography on a chip in the analysis of hemopexin glycopeptides[J].J Chromatogr A, 2017, 1519 (23) :152-155.
[13]Min J Z, Yano H, Matsumoto A, et al.Simultaneous determination of polyamines in human nail as 4- (N, N-dimethylaminosulfonyl) -7-fluoro-2, 1, 3-benzoxadiazole derivatives by nano-flow chip LCcoupled with quadrupole time-of-flight tandem mass spectrometry[J].Clin Chim Acta, 2011, 412 (1-2) :98-106.
[14]Zhu K Y, Leung K W, Ting A K L, et al.The establishment of a highly sensitive method in detecting ketamine and norketamine simultaneously in human hairs by HPLC-Chip-MS/MS[J].Forensic Sci Int, 2011, 208 (1-3) :53-58.
[15]Zhao C X, Wu Z M, Xue G, et al.Ultra-high capacity liquid chromatography chip/quadrupole time-of-flight mass spectrometry for pharmaceutical analysis[J].JChromatogr A, 2011, 1218 (23) :3669-3674.
[16] 中华人民共和国卫生部.GB 5749-2006生活饮用水卫生标准[S].[The Minister of Health of the People's Republic of China.GB 5749-2006 Standards for drinking water quality[S].]

由于具有灵敏度高、检出限低、线性范围宽及多元素可以同时测定的特点,电感耦合等离子体质谱法(ICP-MS)在元素分析的各个领域都得到了广泛应用。当分析复杂基质时,ICP-MS存在基质效应、多原子干扰和离子干扰。一般而言,可以采用内标法[1]、编辑...

抗感冒药种类繁多,但药物成分不尽相同,大多以对乙酰氨基酚、氯苯那敏、右美沙芬、伪麻黄碱、盐酸金刚烷胺等为主要成分,在解热镇痛、抗组胺作用、镇咳祛痰、收缩血管、抗病毒等方面发挥着重要作用。...

酶分子产生于活细胞,其是具有特异识别底物和催化功能性质的蛋白质或RNA分子,亦是一种极其重要的生物催化剂。人体和哺乳动物体内至少含有5 000种酶,在凝血过程、蛋白质代谢、组织再生和免疫防御等许多生物活动中扮演着重要角色。...

润滑油在机械设备和运输工具中起润滑、冷却、散热、密封、抗腐蚀、防锈、清洁、应力分散缓冲、动能传递和绝缘等作用,占整个润滑材料的85%。...