生理学论文

摘 要: 在血液循环系统中,血小板在抑制因子的作用下,处于静息状态。当机体出血或外界因素刺激时,血小板活化,产生聚集、黏附和释放反应,释放出二磷酸腺苷(ADP)、花生四烯酸(AA)、血小板活化因子和5-羟色胺等物质,招募更多的血小板黏附于出血处,从而启动凝血过程,发挥止血作用。当止血反应完成后,血小板发生解聚,恢复到静息状态。然而,在病理条件下,血小板的内在解聚能力下降,形成过度活化的血小板,产生病理性血栓,导致急性缺血性心血管疾病的发生。临床使用抗血小板药物控制血小板的活化,治疗急性缺血性心血管疾病。然而,目前临床上常用的抗血小板药物发挥抗血小板活化作用的同时,影响了血小板正常的生理性止血作用,产生出血等副作用。因此,我们需要研发新型抗血小板药物,使其既能发挥抗血小板作用,又能减少出血等副作用。本文将对血小板负性调控机制进行综述,为进一步研究抗血小板药物提供思路。
关键词: 血小板; 负性调控; 解聚;
Abstract: In normal blood flow circulation, platelets are stimulated by activators, as well as regulated by some inhibitory factors. When the body completes hemostasis, platelets depolymerize and back to resting state. However, under pathological condition, the intrinsic deaggregation ability of platelet decreases, and excessive platelet activation produces pathological thrombus. Therefore, anti-platelet therapy is an important direction for the treatment of ischemic cardiovascular disease. A variety of clinically effective anti-platelet medicines have been developed through platelet activation mechanism, but there are common bleeding adverse reaction at the same time. In this review, we summarized the mechanism of platelet negative regulation to limit platelet excessive activation, in order to provide new idea for further study on anti-platelet drug without bleeding.
Keyword: platelet; negative regulation; depolymerization;
心血管疾病,又称“世界第一杀手”,每年夺去1 000多万人的生命,到2030年死亡人数将增加到2 360万[1]。在中国,五分之二的死亡归因于心血管疾病,而绝大多数的急性心血管疾病与血栓形成有关,如在动脉粥样硬化与心肌梗死中,血小板的激活是该过程的启动因素[2]。血小板是由骨髓巨核细胞的细胞质产生的无核细胞碎片,当血管发生损伤时,内皮基质暴露,血小板与之结合,并产生聚集、变形、黏附和颗粒内容物的释放等反应。血小板释放的颗粒内容物包括血小板活化因子(platelet activating factor,PAF)、花生四烯酸(arachidonic acid,AA)、二磷酸腺苷(adenosine diphosphate,ADP)、凝血酶、肾上腺素和5-羟色胺等物质,这些激动剂与血小板表面相关受体结合,招募血流中更多的血小板到达血管损伤部位,最终启动凝血过程,发挥止血作用[3]。然而,过度的血小板聚集形成病理性血栓使血管闭塞导致缺血性脑卒中、心肌梗死等缺血性心血管疾病的发生。血小板不仅具有聚集的能力,还有内在的解聚功能,血管栓塞性疾病及糖尿病患者的血小板不仅处于亚激活状态,它的内在解聚功能显着降低[4]。血小板通过负性调控,阻断血小板的继续活化,使其解聚,从而防止血栓的过度形成[5]。多种分子和蛋白质对血小板功能具有负性调控作用,综述如下。
1、 一氧化氮
一氧化氮(nitricoxide,NO)是一种中性氧化物,根据来源可分为内源性一氧化氮和外源性一氧化氮。外源性一氧化氮是极不稳定的有毒气体,内源性一氧化氮可为人体利用。内源性一氧化氮由3种不同亚型的一氧化氮合酶(NO synthase,NOS)催化L-精氨酸(L-arginine,L-Arg)产生,细胞外的L-Arg经跨膜转运进入细胞内,经NOS催化生成NO和L-瓜氨酸[6]。NO有高度脂溶性,极易扩散通过生物膜。内源性NO导致血管扩张,抑制血小板在内皮细胞表面黏附和血小板的聚集作用。NO可以结合直接溶于胞质溶胶中的可溶性鸟苷酸环化酶(soluble guanylyl cyclase,sGC)。sGC通过产生环磷酸鸟苷(cGMP)与NO结合,GMP又激活蛋白激酶G(protein kinase G,PKG),进而通过PKG抑制血小板活化[7]。NO又可以刺激腺苷酸环化酶生成环磷酸腺苷(cyclic adenosine monophosphate,cAMP),通过蛋白激酶A(protein kinase A,PKA)抑制血小板聚集。NO还可以减少血小板膜表面糖蛋白(如P-选择素、CD63和GpIIb/IIIa)的表达及颗粒的释放,阻断血小板在损伤内皮细胞处沉积及与纤维蛋白原的结合,从而阻断血小板的活化[8]。NO抑制血小板源性生长因子和血管内皮细胞生长因子,防止内皮下层的平滑肌细胞暴露于这些强效促增殖的物质。
2 、前列环素
前列环素2(prostacyclin 2,PGI2)是前列腺素家族的重要成员,属于内皮源性血管舒张因子,是血栓素的拮抗剂,具有抑制血小板凝集和扩张血管的作用,可延缓血栓的形成,用于肺动脉高压、高血压、动脉粥样硬化和心肌梗死等疾病的治疗。PGI2通过作用于细胞膜上组织特异性的G蛋白偶联受体,即前列环素受体(prostacyclin receptor,IP-R)及相关的信号通路而发挥对心血管系统的调节作用。在血小板膜表面,PGI2通过刺激IP-R发挥抑制血小板活化的功能。激活后的IP-R可以刺激腺苷酸环化酶合成cAMP,后者又激活PKA,可以抑制血小板的活化[9]。NO还可以增强抑制血小板的作用,两者具有协同作用。然而,PGI2和NO都具有非常短的半衰期并且它们的抑制作用是可逆的,可以被浓度较高的激动剂激活的血小板所掩盖。因此,当血小板抑制信号和活化信号的平衡被打破后,血小板的解聚主要由免疫受体酪氨酸抑制基序(immunoreceptor tyrosine-based inhibition motif,ITIM)途径介导的负性调控机制完成(图1)。
3 、免疫受体酪氨酸抑制基序
免疫受体酪氨酸抑制基序ITIM是免疫细胞的抑制性调节分子,如FcRγ、PD-1、CD-22和KIR等,基本结构为I/VxYxxL(I:Ile,V:Val,Y:Tyr,L:Leu)。ITIM位于免疫细胞某些受体分子的胞内段,酪氨酸被Ser活化后可招募并活化带有SH2的PTP,阻断信号转导通路,发挥负向调节作用。ITIM被PTP分子上的SH2结构域所结合,从而招募PTP并使之活化,起到抑制PTK参与的激活信号转导通路作用。
ITIM受体包括血小板内皮细胞黏附分子-1(platelet endothelial cell adhesion molecule-1,PE-CAM-1)、癌胚抗原黏附分子-1/-2(carcinoembryonic antigen cell adhesion molecule-1/-2,CEACAM-1和CEACAM-2)和G6b-B。这些受体在细胞质尾部具有共同的序列,可以与包含非跨膜ptp、SHP-1和SHP-2的SH2结构域结合,导致酪氨酸家族激酶的磷酸化,防止SFKs激活整合素αIIbβ3[10]。酪氨酸激酶Syk和LAT去磷酸化后,还可以通过抑制PLC的磷酸化和PIP3的水解,发挥抑制血小板活化的作用[11]。
图1 血小板负性调控机制图
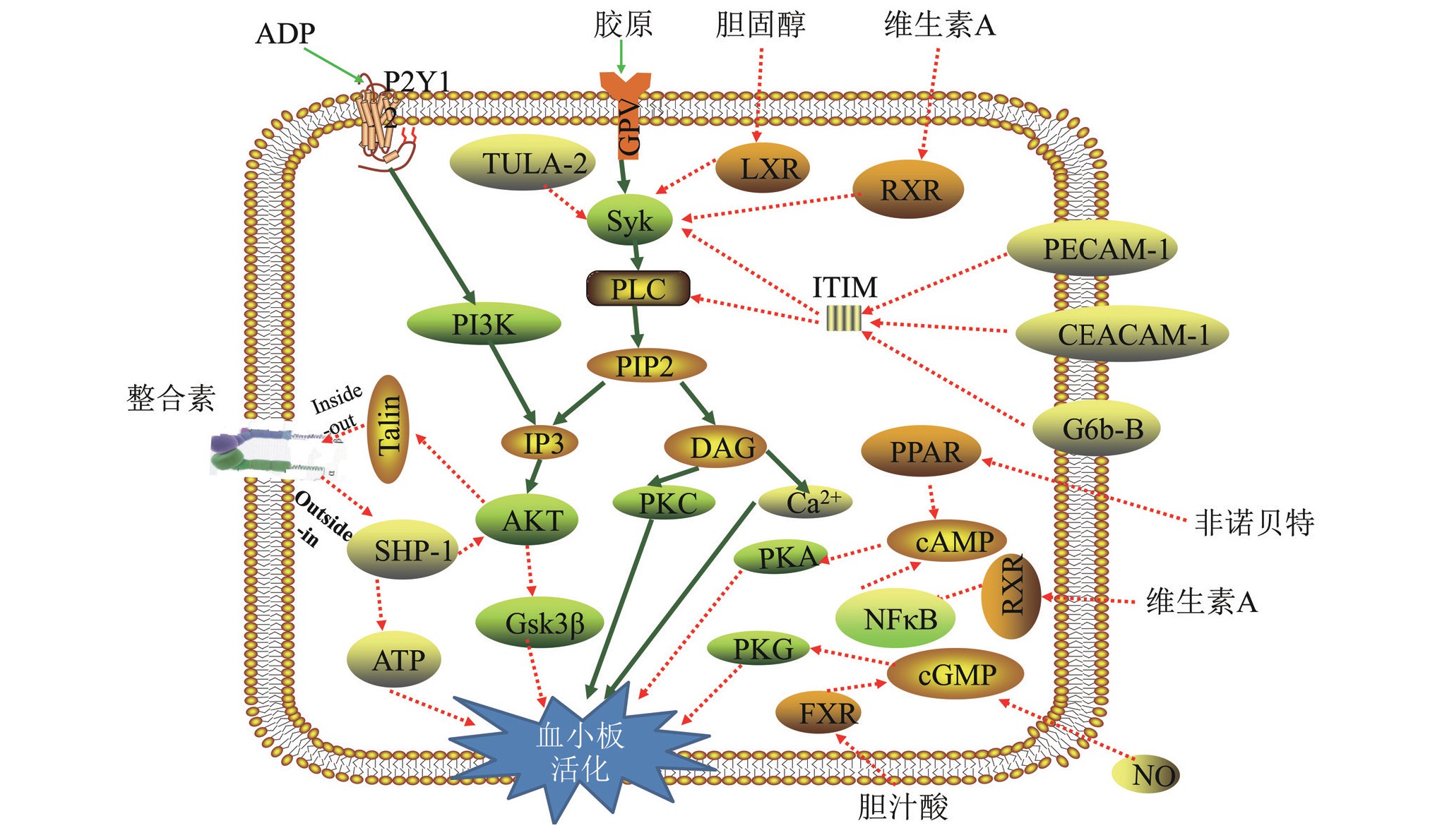
TULA-2:T细胞泛素配体-2;ADP:二磷酸腺苷;PLC:磷脂酶C;PIP2:4,5-磷脂酰肌醇二磷酸;IP3:1,4,5-三磷酸肌醇;DAG:甘油二酯;c AMP:环磷酸腺苷;PI3K:磷脂酰肌醇-3-激酶;AKT:蛋白激酶B;c GMP:环磷酸鸟苷;PPAR:过氧化物酶体增殖物活化受体;FXR:法尼酯X受体;RXR:视黄醇X受体;NFκB:核因子κB;NO:一氧化氮;ITIM:免疫受体酪氨酸抑制基序;PECAM-1:血小板内皮细胞黏附分子-1;PKA:蛋白激酶A;PKG:蛋白激酶G
4 、血小板内皮细胞黏附分子
PECAM-1又称CD31,相对分子质量大约为130 000,属于免疫球蛋白超家族,可与整合素αvβ3结合,主要表达于血小板、白细胞、单核细胞、中性粒细胞、内皮细胞和淋巴细胞,参与吞噬细胞、NK细胞活化T细胞穿越毛细血管壁的过程[12]。PECAM-1由6个免疫球蛋白同源结构域、19个残基单通道跨膜结构域和118个氨基酸细胞质尾组成。PECAM-1在血小板中是其抑制性受体,在血小板负性调控的作用下,PECAM-1活化,其胞内段的Y663和Y686位点激活蛋白酪氨酸激酶(protein tyrosine kinase,PTK),使ITIM磷酸化,成为含SH2结构域的酪氨酸磷酸酶(SHP-1、SHP-2、SHIP)的潜在结合位点[13]。形成PECAM-1/SHP-2复合物,在循环血细胞中可以抑制过多的酪氨酸激酶的激活。SHP-1通过水解PIP2抑制第二信使IP3和DAG的形成,阻断血小板中Ca2+的外流,使Ca2+浓度降低,抑制PKC的活化,从而抑制血小板的活化[14]。
5、 癌胚抗原相关细胞黏附分子
癌胚抗原相关细胞黏附分子-1(carcin embryonic antigen related cellul aradhesion molecule-1,CEA-CAM-1),也被称作CD66a、胆汁糖蛋白(biliarglyeoprotein,BGP)和C-CAM,是一种细胞表面跨膜糖蛋白,也是癌胚抗原家族的一个成员,属于免疫球蛋白超家族黏附分子。CEACAM-1广泛地表达于上皮细胞和血管内皮细胞,具有延缓肿瘤的生长及上皮细胞的增殖、阻止T淋巴细胞的活化、促进血管的形成等多种生物作用。CEACAM-1/-2在鼠和人血小板中均有表达,CEACAM-1与SHP-1以及SHP-2结合,诱导SFK脱磷酸化,非活性SFK不能磷酸化胶原介导的GPVI/FcRγ血小板活化信号通路[15]。CEACAM-2用于负性调节胶原诱导的GPVI-FcRg-链、C-型凝集素样受体-2(C-type lectin like receptor-2,CLEC-2)和诱导的血小板活化信号传导途径。CEACAM-2具有基于ITIM介导的黏附和信号传导特性。CEACAM在血小板活化后易位至细胞表面,并参与否定整合素由内向外的信号通路调节,在CEACAM-2基因敲除小鼠中,血栓形成更加稳定,血小板对胶原诱导血小板的活化更灵敏[16]。
6、 G6b-B
G6b-B是I型跨膜糖蛋白,由单个细胞外免疫球蛋白样可变型结构域、跨膜区和细胞质尾组成。细胞质尾部含有富含脯氨酸的脯氨酸区域、ITIM和C末端ITSM。G6b-B抑制胶原相关肽(collagenrelated peptide,CRP)和二磷酸腺苷(adenosine diphosphate,ADP)诱导的血小板聚集。G6b属于免疫球蛋白超家族,有G6b-A和G6b-B两种亚型,是血小板稳态的关键调节因子。G6b对血小板稳态的调节依赖于蛋白酪氨酸磷酸酶对SHP-1和SHP-2的募集作用,通过阻断底物结合蛋白酪氨酸磷酸酶,从而抑制蛋白酪氨酸磷酸酶对血小板的活化作用。在G6b-B基因敲除小鼠中,巨核细胞的金属蛋白酶浓度升高,促进血小板表面受体GPVI和GPIbα的水解,抑制整合素由内向外的信号传导,阻断血小板活化信号途径[17]。
7 、内皮细胞选择性黏附分子
内皮细胞选择性黏附分子(endothelial cellselective adhesion molecule,ESAM)是一种相对分子质量约55 000的I型膜糖蛋白,具有两个细胞外免疫球蛋白结构域(V型和C2型)。ESAM在巨核细胞和血小板中特异性表达,贮存在α颗粒中。在ESAM基因敲除小鼠中,血小板的最大聚集率和血栓栓块明显增大,在尾部横断后能实现更稳定的止血。尽管聚集增加,但ESAM基因敲除小鼠中血小板在细胞溶质钙动员和αIIbβ3活化方面没有明显变化,可能ESAM依赖相邻细胞分子的同源性发挥其作用[18]。
8 、蛋白酪氨酸磷酸酶途径
T细胞泛素配体-2(T-cell ubiquitin ligand-2,TULA-2)又称为p70、STS-1和UBASH3B,是一种蛋白酪氨酸磷酸酶,在T淋巴细胞、肥大细胞、破骨细胞以及血小板中发挥抑制受体信号传递的作用。TULA-2与Syk的Tyr(P)346位点结合,使其去磷酸化,减弱Syk激酶的活性,抑制GPVI的信号传导,继而下调PLC途径介导的血小板活化途径,抑制Ca2+的释放,降低血小板内钙离子的浓度,减少颗粒释放,抑制血小板的活化[19]。研究发现,在TULTA-2基因敲除小鼠中,小鼠的尾出血时间明显缩短,血栓形成更加稳定[20]。
9、 整合素
整合素(integrin)是α亚基和β亚基以非共价键形式连接组成的异二聚体,是一种跨膜糖蛋白受体,其家族成员在结构上的共同点是由胞外区、跨膜区及胞内区三部分组成[21]。血小板诱导剂作用于血小板表面的非整合素受体后,引起PI3K/AKT的磷酸化,进而使Talin和Kindlins蛋白结合到整合素β亚基的尾部,经Inside-out信号通路活化整合素受体,使得膜表面原本处于静息状态的整合素发生构象改变而被激活。活化的αIIbβ3与血浆中的纤维蛋白原和vWF结合,导致血小板相互聚集成团,形成血小板血栓[22]。随着血小板的活化,整合素αIIbβ3介导的Outside-in的信号导致Talin和Kindlin-3从整合素β3上脱落,促使已经激活的整合素失活。同时整合素Outside-in的信号激活SHIP-1,SHIP-1介导AKT去磷酸化,导致PI3K/AKT信号通路失活。同时,SHIP-1还抑制血小板ATP的释放,由于已经激活的整合素发生失活,又没有新的释放物刺激整合素再次激活,所以已经聚集的血小板发生解聚。当低剂量的弱诱导剂ADP诱导时,整合素αIIbβ3的outside-in信号通路可以产生负调控信号抑制血小扳聚集,而这种负调控信号在大剂量诱导剂或强诱导剂的作用下会被正调控信号所掩盖[23]。
1 0 、核受体
核受体(nuclear receptor,NR)是细胞核内的配体依赖性的转录因子,众多生理学过程需要内分泌信号与人体内的受体进行协同调节,如:雄激素受体(androgen receptor,AR)、孕激素受体(progesterone receptor,PR)和雌激素受体(estrogen receptor,ER)在生殖中起重要作用;糖皮质激素受体(glucocorticoid receptor,GR)在葡萄糖代谢和炎症中起作用;甲状腺激素受体(thyroid hormone receptor,TR)在有氧代谢方面起作用;过氧化物酶体增殖物活化受体(peroxisome proliferator-activated receptor,PPAR)调节脂质和能量代谢等。核受体通过基因组和非基因组两种方式发挥作用,而血小板中的核受体通过非基因组的方式发挥作用。尽管血小板是无核细胞,但含有mRNA、rRNA、tRNA、miRNA、核受体及核因子NFκB,这些与基因转录有关的因子和RNA都来源于血小板脱落时的巨核细胞[24]。血小板中的核受体主要通过升高cAMP和cGMP的浓度和负性调控GPVI信号途径抑制血小板活化。
肝X受体(liver X receptor,LXR)与配体结合后,一方面促进血小板变形、颗粒释放以及磷脂酰丝氨酸暴露,使血小板处于促凝状态;另一方面使整合素头部弯曲、处于静息状态,并去磷酸化酪氨酸激酶LAT和Syk,减弱PLC对PIP2的水解作用,降低第二信使DAG和IP3的浓度,促进Ca2+外流,抑制PKC对血小板的活化作用,最终使血小板处于抑制状态。
法尼酯X受体(farnesoid X receptor,FXR)升高cGMP的浓度,激活PKG,而PPAR升高cAMP的浓度,激活PKA,抑制血小板活化。FXR配体与受体结合,作用于核因子NFκB,通过PKA抑制血小板活化。维甲酸受体家族是其中的重要组成部分。维甲酸核受体又可分为视黄酸受体(retinoid acid receptor,RAR)和视黄醇X受体(retinoid X receptor,RXR),均包括α、β、γ三种亚型。已有文献报道[25],RXR与相应配体(如贝沙罗汀、9-顺式视黄酸、二十二碳六烯酸和甲戊二酸等)结合后,作用于核因子NFκB,激活PKA。PKA是蛋白激酶A,其活性受到细胞内cAMP浓度水平的调节,并且可通过底物GPIbβSer166的磷酸化程度来表示PKA的活化程度。PKA还可以抑制PLC的磷酸化和改变整合素的构型,促进血小板解聚,使血小板恢复到静息水平[26]。核因子NFκB是一种调节因子,在炎症和血管损伤时发挥重要作用。核因子NFκB不仅能通过PKA抑制血小板活化和血栓形成,还可以促进血小板膜糖蛋白(platelet glycoprotein Ib,GPIbα)的脱落,阻断血小板活化信号的传递[27]。核受体包括PPAR、LXR和FXR等配体,具有抗血小板活化和动脉粥样硬化的作用。这些配体提供了一种潜在的新型治疗模式,可以靶向抗血栓和改善其他一系列病理生理条件[28]。
1 1、 LIM-Domain家族
LIM结构域(LIM-Domain)蛋白家族一共有7个成员,具有相似的结构和生物学功能。LIM结构域家族成员包括CLP36、Paxillin、Hic-5和leupaxin,都与血小板激活有关。Hic-5主要表达于人血小板;CLP36、Paxillin和leupaxin在人和鼠血小板均有表达。CLP相对分子质量为38 000,N端由一个PDZ结构域组成,C端由3个LIM结构域组成。CLP36基因敲除小鼠的血小板可由CRP、胶原蛋白和convulxin诱导,而ADP和凝血酶并不能诱导血小板活化。CLP36负性调节血小板活化主要与抑制GPVI-ITAM-PLCγ2信号通路有关。在CLP36基因敲除小鼠中,GPVI介导的血小板活化途径中PLC的活性增强,增加Ca2+的释放和Ca2+流入,从而诱导更快的α-颗粒和致密颗粒的释放以及整合素的激活[29]。
Paxillin是一种LIM结构域蛋白,最初被鉴定为癌基因v-src的底物[27]。Paxillin敲除小鼠中血小板略微增大,且其血小板黏附能力增强、体内血栓形成增加、TXA2的合成和致密颗粒的释放也得到了增强。Paxillin还可以通过抑制整合素αIIbβ3的活化发挥抑制血小板聚集的作用[30]。
1 2、 其它血小板负性调节因子
磷脂酶D(phospholipase D,PLD)催化磷脂酰胆碱水解成磷脂酸(phosphatidic acid,PA)和胆碱,PLD参与广泛的细胞生物学过程,包括脱颗粒、内吞作用、细胞侵袭和细胞骨架重组[31]。此外,PLD还可以调节肿瘤发生、癌细胞的存活和侵袭。PLD具有两种亚型PLD1和PLD2,都含有N端氨基酸和C端氨基酸,C端氨基酸对PLD的正确折叠影响较大,N端氨基酸对其催化反应的作用较为明显[32]。PLD1可被PKC和小GTP酶激活。血小板中缺乏PLD1会抑制整合素αIIbβ3的活化和血栓的形成,但脱颗粒和聚集作用不受影响。PLD2是血小板敏感型的潜在负调节因子,PLD2被多种激动剂激活,如胶原蛋白、凝血酶和TXA2。丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)属于丝氨酸/苏氨酸蛋白激酶家族,包括三个主要亚组:ERK、p38MAPK和JNK。ERK、p38MAPK和JNK1存在于血小板中,由各种激动剂激活,并参与血小板颗粒的释放、聚集、黏附和血栓形成。研究表明,PLD通过MAPK和AKT途径抑制血小板活化、颗粒释放和随后的血小板聚集[33]。
Prx II(peroxiredoxin II)是一种细胞过氧化物酶,可分解血小板内的过氧化氢(H2O2)。H2O2具有促进GPVI信号激活的作用。PrxII/SHP-2介导的抑制血小板活化发生在脂筏中。PrxII主要通过下调GPVI的信号传导抑制血小板的活化[34]。Prx II缺陷型血小板在体外显示出黏附和聚集活性功能的增强。
适配器蛋白2(adapter protein disabled 2,DAB2)包含三个结构域:N末端磷酸酪氨酸结合(PTB)结构域、天冬氨酸-脯氨酸-苯丙氨酸(DPF)基序和C末端富含脯氨酸的区域。人的DAB2基因位于染色体5p13位,DAB2来源于血小板活化后的致密颗粒中,DAB2可以触发多种生物学反应,参与胞吞作用和抑制血小板的聚集反应。DAB2的RGD结构域通过磷酸化作用与整合素αIIbβ3的胞外区结合,竞争整合素与纤维蛋白原的结合,进而抑制血小板的聚集。DAB2基因敲除小鼠中,凝血酶诱导血小板聚集,栓块收缩,并在纤维蛋白原上扩散,伸出伪足,形成血小板聚集体[35]。
硫氧还蛋白相关跨膜蛋白1(thioredoxin-related transmembrane protein 1,TMX1)是一种蛋白二硫异构酶,属于胞外血小板抑制剂,也是第一个负性调控血小板功能的二硫异构酶。TMX1主要通过二硫键的作用使整合素处于静息状态,从而抑制血小板聚集和ATP的释放,发挥抗血小板活化的作用[36]。
目前临床上常用的抗血小板药物如氯吡格雷、阿西单抗、阿司匹林等,具有强大的抗血小板聚集作用,但同时也易产生出血等副作用。一方面抗血小板药物影响了血小板的正常生理止血作用;另一方面,在血栓栓塞性疾病中,由血小板负性调控机制介导的解聚功能下降,不能使血小板恢复至静息状态,进一步增加了出血的风险。健康血管脉络系统释放出NO和PGI2,使血小板处于抑制状态;当血小板活化后,内源性负性调控机制能够控制血小板的过度活化,使血小板处于静息状态。因此,我们可以通过研究血小板的负性调控机制,探讨其潜在的治疗意义,从血小板解聚的角度出发,研发出新型防治血栓、同时又能降低出血风险的药物[37]。
参考文献
[1] Nguyen TH, Palankar R, Bui VC, et al. Rupture forces among human blood platelets at different degrees of activation. Sci Rep, 2016, 5(6):25402-25414
[2] Barrett TJ, Schlegel M, Zhou F, et al. Platelet regulation of myeloid suppressor of cytokine signaling 3 accelerates atherosclerosis. Sci Transl Med, 2019, 11(517):481-492
[3] Stalker TJ, Newman DK, Ma P, et al. Platelet signaling.Handb Exp Pharmacol, 2012, 3(210):59-85
[4] Gligorijevic N, Robajac D, Nedic O. Enhanced platelet sensitivity to IGF-1 in patients with type 2 diabetes mellitus. Biochemistry, 2019, 84(10):1213-1219
[5] Bye AP, Unsworth AJ, Gibbins JM. Platelet signaling:a complex interplay between inhibitory and activatory networks. J Thromb Haemost, 2016, 14(5):918-930
[6] Radomski MW, Palmer RM, Moncada S. An L-arginine/nitric oxide pathway present in human platelets regulates aggregation. J Natl Acad, 1990, 87(13):5193-5197
[7] Smolenski A. Novel role of cAMP/cGMP-dependent signaling in platelets. J Thromb Haemost, 2012, 10(2):167-176
[8] Dangel O, Mergia E, Karlisch K, et al. Nitric oxide-sensitive guanylyl cyclase is the only nitric oxide receptor mediating platelet inhibition. J Thromb Haemost, 2010,8(6):1343-1352
[9] Lv L, Jiang SS, Xu J, et al. Protective effect of ligustrazine against myocardial ischaemia reperfusion in rats:the role of endothelial nitric oxide synthase. Clin Exp Pharmacol Physiol, 2012, 39(1):20-27
[10] Walsh MT, Kinsella BT. Regulation of the human prostanoid TPαand TPβreceptor isoforms mediated through activation of the EP1 and IP receptors. Br J Pharmacol,2000, 131(3):601-609
[11] Coxon CH, Geer MJ, Senis YA. ITIM receptors:more than just inhibitors of platelet activation. Blood, 2017,129(26):3407-3418
[12] Tourdot BE, Brenner MK, Keough KC, et al. Immunoreceptor tyrosine-based inhibitory motif(ITIM)-mediated inhibitory signaling is regulated by sequential phosphorylation mediated by distinct nonreceptor tyrosine kinases:a case study involving PECAM-1. Biochemistry, 2013,52(15):2597-2608
[13] Newman PJ, Newman DK. Signal transduction pathways mediated by PECAM-1:new roles for an old molecule in platelet and vascular cell biology. Arterioscler Thromb Vasc Biol, 2003, 23(6):953-964
[14] Moraes LA, Barrett NE, Jones CI, et al. Platelet endothelial cell adhesion molecule-1 regulates collagen-stimulated platelet function by modulating the association of phosphatidylinositol 3-kinase with Grb-2-associated binding protein-1 and linker for activation of T cells. J Thromb Haemost, 2010, 8(11):2530-2541
[15] Hu M, liu P, Liu Y, et al. Platelet Shp2 negatively regulates thrombus stability under high shear stress. J Thromb Haemost, 2019, 17(1):220-231
[16] Wong C, Liu Y, Yip J, et al. CEACAM1 negatively regulates platelet-collagen interactions and thrombus growth in vitro and in vivo. Blood, 2009, 113(8):1818-1828
[17] Stalker TJ, Wu J, Morgans A, et al. Endothelial cell specific adhesion molecule(ESAM)localizes to platelet-platelet contacts and regulates thrombus formation in vivo. J Thromb Haemost, 2009, 7(11):1886-1896
[18] Coxon CH, Sadler AJ, Huo J, et al. An investigation of hierachical protein recruitment to the inhibitory platelet receptor, G6B-b. PLoS One, 2012, 7(11):49543-49553
[19] Reppschl?ger K, Gosselin J, Dangelmaier CA, et al.TULA-2 protein phosphatase suppresses activation of Syk through the GPVI platelet receptor for collagen by dephosphorylating Tyr(P)346, a regulatory site of Syk. J Biol Chem, 2016, 291(43):22427-22441
[20] Kostyak JC, Mauri BR, Dangelmaier C, et al. TULA-2 Deficiency enhances platelet functional responses to CLEC-2 agonists. TH Open, 2018. 2(4):411-419
[21] Shen B, Zhao X, O’Brien KA, et al. A directional switch of integrin signaling and a new anti-thrombotic strategy.Nature, 2013, 503(7474):131-145
[22] Joo SJ. Mechanisms of platelet activation and integrin alphaIIbeta3. Korean Circ J, 2012, 42(5):295-301
[23] Dai B, Wu P, Xue F, et al. Integrin-αIIbβ3-mediated outside-in signalling activates a negative feedback pathway to suppress platelet activation. J Thromb Haemost, 2016,116(05):918-930
[24] Unsworth AJ, Flora GD, Gibbins JM. Non-genomic effects of nuclear receptors:insights from the anucleate platelet. Cardiovasc Res, 2018, 114(5):645-655
[25] Amanda J, Unsworth, Gagan D. RXR ligands negatively regulate thrombosis. Arterioscler Thromb Vasc Biol,2017, 37(5):812-822
[26] Gambaryan S, Kobsar A, Rukoyatkina N, et al. Thrombin and collagen induce a feedback inhibitory signaling pathway in platelets involving dissociation of the catalytic subunit of protein kinase A from an NFkappaB-IkappaB complex. J Biol Chem, 2010, 285(24):18352-18363
[27] Fuentes EA, Rojas, Palomo I. NF-kappaB signaling pathway as target for antiplatelet activity. Blood Rev, 2016,30(4):309-315
[28] Evans RM, Mangelsdorf DJ. Nuclear receptors, RXR,and the big bang. Cell, 2014, 157(1):255-266
[29] Gupta S, Braun A, Morowski M, et al. CLP36 is a negative regulator of glycoprotein VI signaling in platelets.Circ Res, 2012, 111(11):1410-1420
[30] Gao J, Huang M, Lai J, et al. Kindlin supports platelet integrin alphaIIbbeta3 activation by interacting with paxillin. J Cell Sci, 2017, 130(21):3764-3775
[31] Sakata A, Ohmori T, Nishimura S, et al. Paxillin is an intrinsic negative regulator of platelet activation in mice.Thromb J, 2014, 12(1):1-14
[32] Zhang H, Chu W, Sun J, et al. Combining cell surface display and DNA shuffling technology for directed evolution of streptomyces phospholipase D and synthesis of phosphatidylserine. J Agric Food Chem, 2019, 10(11):5394-5463
[33] Lu WJ, Chung CL, Chen RJ, et al. An antithrombotic strategy by targeting phospholipase D in human platelets.J Clin Med, 2018, 7(11):440-455
[34] Jang JY, Wang SB, Min JH, et al. Peroxiredoxin II is an antioxidant enzyme that negatively regulates collagen-stimulated platelet function. J Biol Chem, 2015,290(18):11432-11442
[35] Tsai HJ, Tseng CP. The adaptor protein Disabled-2:new insights into platelet biology and integrin signaling.Thromb J, 2016, 14(1):28-35
[36] Zhao Z, Wu Y, Zhou J, et al. The transmembrane protein disulfide isomerase TMX1 negatively regulates platelet responses. Blood, 2019, 133(3):246-251
[37] Stefanini L, Bergmeier W. Negative regulators of platelet activation and adhesion. J Thromb Haemost, 2018, 16(2):220-230

在漫长的生命发展历程中,地球上的各种生物包括人类在内,其行为和生理功能都表型出一定的节律性。阳春三月,北半球的候鸟纷纷北飞;秋令时刻,鸿雁频频南飞;没有闹钟铃声的人们依然每天能够如期觉醒。这些有规律的周期现象说明各种生物不止是知道时间,乃...

本文主要从有氧运动对患病群体和中老年群体血流变影响展开综述, 旨在对有氧运动与血流变的关系进行梳理分析, 为该方面的研究提供文献参考。...
间套作在没有扩大土地面积的前提下提高了粮食产量,为解决世界人口的温饱问题做出了重要贡献。研究表明,间套作种植能够有效提高资源利用率和粮食产量,增强农业系统的抗风险能力,增加水土保持能力,提高土壤肥力,同时能够抑制病虫草害的发生,是一种基于...
1前言哺乳动物对不同环境的适应及其调节机理是生理生态学研究的热点之一。生态代谢理论是近年来生态学领域的重要进展,不仅有效地解释了生物的代谢率随个体大小和温度的变化规律,还通过代谢率将个体、种群、群落、生态系统水平上的生物学模式联系起来(Mee...

骨骼是一个动态器官,机体通过对骨形成与骨吸收的精密调控,重塑骨骼。成骨细胞和破骨细胞是参与骨重建的主要细胞,分别介导骨形成与骨吸收过程,维持骨代谢动态平衡,使骨骼保持完整的结构和一定的强度。...
社会交往是个体在社会群体里必不可少的沟通方式,有助于个体成长和后代繁衍。即使简单的社会交往行为也需要神经系统精确的分析、整合外界信息并调节运动系统使行为正常进行。许多精神-神经性疾病(如孤独症、抑郁症等)的患者都表现出不同程度的社会交往障...
兔尿生成的影响因素实验是生理学泌尿系统的经典实验,此实验为生理学实验教学中的必做内容之一。尿生成的过程包括:肾小球的滤过作用、肾小管与集合管的重吸收作用以及分泌作用。凡是影响这些过程的因素,都会因影响尿生成从而引起尿量变化。因葡萄糖有...
国家自然科学基金委员会生命科学部、医学科学部与政策局于2014年11月24~26日在南京联合举办了第128期双清论坛。本次论坛的主题为生殖生物学研究的挑战和对策,重点关注动物和人的生殖,论坛执行主席由中国科学院上海生命科学研究院张永莲院士、南京医科大...
佳木斯舞步健身操是人们非常喜欢的一项健身运动项目。为了激发人们参加佳木斯舞步健身操的积极性,更好的体现佳木斯舞步健身操的锻炼价值,本实验对忻州师范学院30名超重女大学生进行13周(2012年3月12日至2012年6月9日)的训练,将实验后实际测得的数据和实...

卷积神经网络(convolutional neural networks, CNN)已被广泛地应用于很多研究领域,如图像分类、人脸识别、交通标志识别等。本文将卷积神经网络用于生物数据核小体DNA序列的识别。...