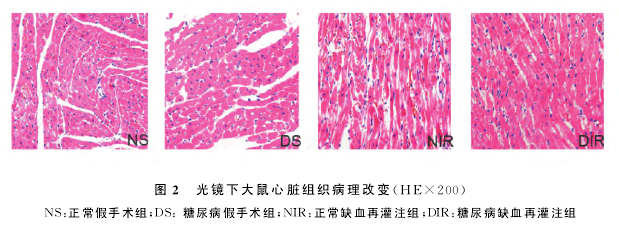
2.3 心肌组织病理改变 显微镜下HE病理结果,NS组心肌纤维排列整齐,心肌细胞无肿胀,横纹清楚;DS组心肌纤维排列稍紊乱,有部分溶解断裂,间质纤维化;NIR组心肌纤维增生、水肿,灶状坏死、断裂,心肌间质充血水肿伴大量炎性细胞浸润;DIR组心损伤较NIR更加严重,见图2.
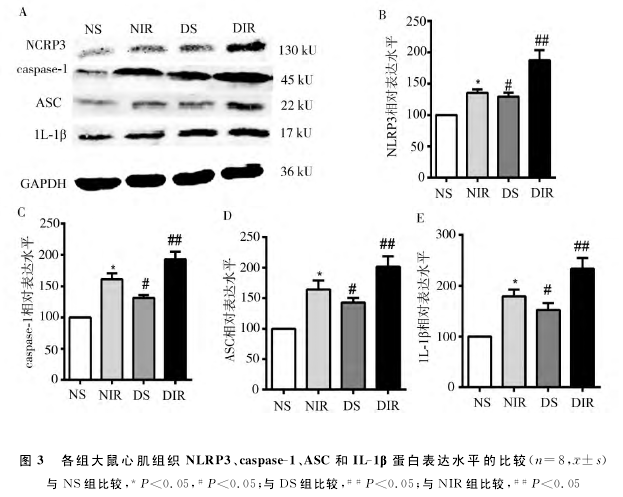
2.4 NLRP3、caspase-1、ASC和IL-1β蛋白表达 与正常大鼠相比,糖 尿 病 大 鼠 心 肌 组 织NLRP3、caspase-1、ASC和IL-1β蛋白表达增加(P<0.05);相比于假手术组,心肌缺血再灌注大鼠心肌组织NLRP3、caspase-1、ASC和IL-1β蛋 白 表 达 增 加(P<0.05);且糖尿病大鼠心肌缺血再灌注时,心肌组织NLRP3、caspase-1、ASC和IL-1β蛋白表达比非糖尿病大鼠缺血再灌注时增加(P<0.05);结果见图3.
3 讨论。
本研究采用腹腔注射链脲佐菌素制备大鼠糖尿病模型,成功制备大鼠糖尿病模型后,普饲8周,确保糖尿病心肌病变形成,此阶段糖尿病心肌病变明显,对心肌缺血再灌注更加敏感[7].采用结扎LAD法制备心肌I/R损伤模型。结果表明,与假手术组比较,非糖尿病大鼠和糖尿病大鼠心肌I/R后缺血和梗死面积明显增加,血清CK-MB和LDH的活性升高;光镜下心肌纤维增生水肿,灶状坏死,断裂,间质充血水肿伴大量炎性细胞浸润,证明成功制备了大鼠心肌缺血再灌注损伤模型。
研究证明,细胞焦亡主要的特点是由caspase-1受胞内多蛋白复合物炎性小体的调控而活化介导的。炎性小体通常是含NACHT(nucleotide bind-ing oligomerization domain)、LRR(leucine rich re-peat)和PYD(pyrin domain)结构域的蛋白、凋亡相关斑点样蛋白(ASC)和caspase-1前体所构成的多蛋白复合物。细胞在外源性相关病原体分子模式(PAMP)和内源性危险相关的分子模式(DAMPS)作用下,通过不同信号途径作用于炎性小体,NLRs暴露出 效 应 结 构 域,通 过CARD-CARD或PYD-PYD同型相互作用,间接或直接通过ASC募集、激活caspase-1,作用于下游因子IL-1β、IL-18前体裂解及释放,介导细胞渗透性肿胀破裂,形成细胞膜小孔,胞内物质(如乳酸脱氢酶LDH等)流出,并诱导其他炎性因子、黏附分子等的合成和释放,放大局部和全身炎症反应,导致细胞促炎程序性死亡-细胞焦亡[8].NOD样 受 体 (NLRP1,NLRP3,NLRC4,NLRC5),PYRIN和AIM2是炎性小体家族的主要成员,在炎症反应发生和维持中起重要作用。NL-RP3炎性小体是目前研究最广泛的NOD样受体,NLRP3炎性识别非微生物危险信号,非生物危险信号如过量的ATP、葡萄糖、活性氧(ROS)、胆固醇和结晶可以激活NLRP3,NLRP3在各种疾病条件下识别非微生物危险信号并导致的非细菌性炎性反应。研究证实,NLRP3在糖尿病心肌病、心肌梗死和心肌缺血再灌注损伤中起重要作用[9,10].
本研究结果表明,糖尿病大鼠相比于非糖尿病大鼠,心 肌 发 生 病 理 改 变,且NLRP3、caspase-1、ASC、IL-1β蛋白表达增加,提示糖尿病状态下,NL-RP3活 性 增 加,通 过 与ASC形 成 炎 性 小 体 激 活caspase-1,caspase-1作用于下游细胞因子IL-1β诱导炎症反应,诱导心肌细胞焦亡发生,导致糖尿病心肌病理损伤。本研究结果显示,I/R显着增加了糖尿病和非糖尿病心肌缺血和梗死面积,并增加CK-MB、LDH活 性,提 示 心 肌 缺 血 再 灌 注 损 伤;NLRP3、caspase-1、ASC和IL-1β蛋白表达都明显增加,提示在心肌缺血再灌注时,氧化应激等刺激因素激活NLRP3炎性体,通过作用于caspase-1,介导心肌细胞焦亡,诱发并加重心肌缺血再灌注损伤。与NIR组比较,DIR组心肌梗死面积增加、CK-MB和LDH活性增加,表明糖尿病相比于非糖尿病大鼠心肌缺血再灌注损伤加重;DIR组的NLRP3、caspase-1、ASC和IL-1β蛋白增加显着,提示糖尿病状态下心肌缺血再灌注时,内源性和外源性损伤刺激通过激活NLRP3炎性小体相关通路作用于caspase-1,介导心肌细胞焦亡,诱发和加重心肌细胞再灌注损伤。
综上所述,本研究结果说明细胞焦亡与糖尿病大鼠心肌病有重要关系,在糖尿病心肌缺血再灌注时,氧化应激等刺激通过NLRP3和ASC炎性小体激活caspase-1信号通路诱导细胞焦亡,使糖尿病心肌对缺血再灌注更加敏感、心肌缺血再灌注损伤加重,在缺血再灌注损伤中起重要作用。通过进一步对糖尿病心肌缺血再灌注损伤的细胞焦亡信号通路的研究,探究细胞焦亡的具体作用机制可以为防治糖尿病心肌缺血再灌注损伤提供新方向和领域。
参考文献:
[1]Keller PF,Carballo D,Roffi M.Diabetes and acutecoronary syndrome[J].Minerva Med,2010,101(2):81-104.
[2]Liu Y,Yang H,Liu LX,et al.NOD2contributes tomyocardial ischemia/reperfusion injury by regulatingcardiomyocyte apoptosis and inflammation[J].Life Sci,2016,149:10-17.
[3]Doitsh G,Galloway NL,Geng X,et al.Cell death bypyroptosis drives CD4T-cell depletion in HIV-1infec-tion[J].Nature,2014,505(7 484):509-514.
[4]Li X,Du N,Zhang Q,et al.MicroRNA-30dregulatescardiomyocyte pyroptosis by directly targeting foxo3ain diabetic cardiomyopathy[J].Cell Death Dis,2014,5:e1479.
[5]Luo B,Li B,Wang W,et al.NLRP3Gene SilencingAmeliorates DiabeticCardiomyopathy in a Type 2Dia-betes Rat Model[J].PLoS One,2014,9(8):e104771.
[6]Yang JR,Yao FH,Zhang JG,et al.Ischemia-reperfu-sion induces renal tubule pyroptosis via the CHOP-caspase-11pathway[J].Am J Physiol Renal Physiol,2014,306(1):F75-F84.
[7]Li H,Liu Z,Wang J,et al.Susceptibility to myocar-dial ischemia reperfusion injury at early stage of type 1diabetes in rats[J]. Cardiovasc Diabetol,2013,12:133.
[8]Byrne BG,Dubuisson JF,Joshi AD,et al.Inflammasomecomponents coordinate autophagy and pyroptosis asmacrophage responses to infection[J].M Bio,2013,4(1):e00620.
[9]Sandanger,Ranheim T,Vinge LE,et al.The NL-RP3inflammasome is up-regulated in cardiac fibro-blasts and mediates myocardial ischaemia-reperfusioninjury[J].Cardiovasc Res,2013,99(1):164-174.
[10]Takahashi M.NLRP3inflammasome as a novel playerin myocardial infarction[J].Int Heart J,2014,55(2):101-105.